Zhuyifan Ye, Nannan Wang, Jiantao Zhou, Defang Ouyang
{"title":"通过耦合生成式对抗网络和图卷积网络预测有机晶体结构","authors":"Zhuyifan Ye, Nannan Wang, Jiantao Zhou, Defang Ouyang","doi":"10.1016/j.xinn.2023.100562","DOIUrl":null,"url":null,"abstract":"<p>Organic crystal structures exert a profound impact on the physicochemical properties and biological effects of organic compounds. Quantum mechanics (QM) based crystal structure predictions (CSP) have somewhat alleviated the dilemma that experimental crystal structure investigations struggle to conduct complete polymorphism studies, but the high computing cost poses a challenge to its widespread application. The current study aims to construct DeepCSP, a feasible pure machine learning framework for minute-scale rapid organic crystal structure prediction. Initially, based on 177,746 data entries from the Cambridge Crystal Structure Database (CSD), a generative adversarial network was built to conditionally generate trial crystal structures under selected feature constraints for the given molecule. Simultaneously, a graph convolutional attention network was employed to predict the density of stable crystal structures for the input molecule. Subsequently, the distances between the predicted density and the definition-based calculated density would be considered as the crystal structure screening and ranking basis, and finally, the density-based crystal structure ranking would be output. Such two distinct algorithms, performing the generation and ranking functionalities respectively, collectively constitute the DeepCSP, which has demonstrated compelling performance in marketed drug validations, achieving an accuracy rate exceeding 80% and hit rate surpassing 85%. Inspiringly, the computing speed of the pure machine learning methodology demonstrates the potential of artificial intelligence in advancing CSP research.</p>","PeriodicalId":36121,"journal":{"name":"The Innovation","volume":"569 1","pages":""},"PeriodicalIF":33.2000,"publicationDate":"2024-01-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Organic crystal structure prediction via coupled generative adversarial networks and graph convolutional networks\",\"authors\":\"Zhuyifan Ye, Nannan Wang, Jiantao Zhou, Defang Ouyang\",\"doi\":\"10.1016/j.xinn.2023.100562\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Organic crystal structures exert a profound impact on the physicochemical properties and biological effects of organic compounds. Quantum mechanics (QM) based crystal structure predictions (CSP) have somewhat alleviated the dilemma that experimental crystal structure investigations struggle to conduct complete polymorphism studies, but the high computing cost poses a challenge to its widespread application. The current study aims to construct DeepCSP, a feasible pure machine learning framework for minute-scale rapid organic crystal structure prediction. Initially, based on 177,746 data entries from the Cambridge Crystal Structure Database (CSD), a generative adversarial network was built to conditionally generate trial crystal structures under selected feature constraints for the given molecule. Simultaneously, a graph convolutional attention network was employed to predict the density of stable crystal structures for the input molecule. Subsequently, the distances between the predicted density and the definition-based calculated density would be considered as the crystal structure screening and ranking basis, and finally, the density-based crystal structure ranking would be output. Such two distinct algorithms, performing the generation and ranking functionalities respectively, collectively constitute the DeepCSP, which has demonstrated compelling performance in marketed drug validations, achieving an accuracy rate exceeding 80% and hit rate surpassing 85%. Inspiringly, the computing speed of the pure machine learning methodology demonstrates the potential of artificial intelligence in advancing CSP research.</p>\",\"PeriodicalId\":36121,\"journal\":{\"name\":\"The Innovation\",\"volume\":\"569 1\",\"pages\":\"\"},\"PeriodicalIF\":33.2000,\"publicationDate\":\"2024-01-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Innovation\",\"FirstCategoryId\":\"95\",\"ListUrlMain\":\"https://doi.org/10.1016/j.xinn.2023.100562\",\"RegionNum\":1,\"RegionCategory\":\"综合性期刊\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MULTIDISCIPLINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Innovation","FirstCategoryId":"95","ListUrlMain":"https://doi.org/10.1016/j.xinn.2023.100562","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
Organic crystal structure prediction via coupled generative adversarial networks and graph convolutional networks
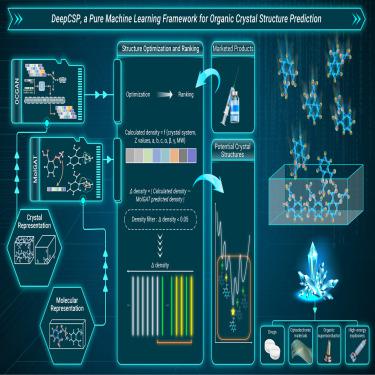
Organic crystal structures exert a profound impact on the physicochemical properties and biological effects of organic compounds. Quantum mechanics (QM) based crystal structure predictions (CSP) have somewhat alleviated the dilemma that experimental crystal structure investigations struggle to conduct complete polymorphism studies, but the high computing cost poses a challenge to its widespread application. The current study aims to construct DeepCSP, a feasible pure machine learning framework for minute-scale rapid organic crystal structure prediction. Initially, based on 177,746 data entries from the Cambridge Crystal Structure Database (CSD), a generative adversarial network was built to conditionally generate trial crystal structures under selected feature constraints for the given molecule. Simultaneously, a graph convolutional attention network was employed to predict the density of stable crystal structures for the input molecule. Subsequently, the distances between the predicted density and the definition-based calculated density would be considered as the crystal structure screening and ranking basis, and finally, the density-based crystal structure ranking would be output. Such two distinct algorithms, performing the generation and ranking functionalities respectively, collectively constitute the DeepCSP, which has demonstrated compelling performance in marketed drug validations, achieving an accuracy rate exceeding 80% and hit rate surpassing 85%. Inspiringly, the computing speed of the pure machine learning methodology demonstrates the potential of artificial intelligence in advancing CSP research.
期刊介绍:
The Innovation is an interdisciplinary journal that aims to promote scientific application. It publishes cutting-edge research and high-quality reviews in various scientific disciplines, including physics, chemistry, materials, nanotechnology, biology, translational medicine, geoscience, and engineering. The journal adheres to the peer review and publishing standards of Cell Press journals.
The Innovation is committed to serving scientists and the public. It aims to publish significant advances promptly and provides a transparent exchange platform. The journal also strives to efficiently promote the translation from scientific discovery to technological achievements and rapidly disseminate scientific findings worldwide.
Indexed in the following databases, The Innovation has visibility in Scopus, Directory of Open Access Journals (DOAJ), Web of Science, Emerging Sources Citation Index (ESCI), PubMed Central, Compendex (previously Ei index), INSPEC, and CABI A&I.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


