{"title":"对生酮饮食耐受的 GLUT-1DS:从临床特征到硅学分析。典型病例报告及文献综述。","authors":"Raffaele Falsaperla, Vincenzo Sortino, Giovanna Vitaliti, Grete Francesca Privitera, Martino Ruggieri, Gaia Fusto, Xena Giada Pappalardo","doi":"10.1007/s10048-023-00742-8","DOIUrl":null,"url":null,"abstract":"<p><p>Glucose transporter type 1 deficiency syndrome (GLUT-1DS) is characterized by alterations in glucose translocation through the blood-brain barrier (BBB) due to mutation involving the GLUT-1 transporter. The fundamental therapy is ketogenic diet (KD) that provide an alternative energetic substrate - ketone bodies that across the BBB via MCT-1 - for the brain. Symptoms are various and include intractable seizure, acquired microcephalia, abnormal ocular movement, movement disorder, and neurodevelopment delay secondary to an energetic crisis for persistent neuroglycopenia. KD is extremely effective in controlling epileptic seizures and has a positive impact on movement disorders and cognitive impairment. Cases of KD resistance are rare, and only a few of them are reported in the literature, all regarding seizure. Our study describes a peculiar case of GLUT-1DS due to a new deletion involving the first codon of SLC2A1 gene determining a loss of function with a resistance to KD admitted to hospital due to intractable episodes of dystonia. This patient presented a worsening of symptomatology at higher ketonemia values but without hyperketosis and showed a complete resolution of symptomatology while maintaining low ketonemia values. Our study proposes an in-silico genomic and proteomic analysis aimed at explaining the atypical response to KD exhibited by our patient. In this way, we propose a new clinical and research approach based on precision medicine and molecular modelling to be applied to patients with GLUT-1DS resistant to first-line treatment with ketogenic diet by in silico study of genetic and altered protein product.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":" ","pages":"69-78"},"PeriodicalIF":1.2000,"publicationDate":"2024-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"GLUT-1DS resistant to ketogenic diet: from clinical feature to in silico analysis. An exemplificative case report with a literature review.\",\"authors\":\"Raffaele Falsaperla, Vincenzo Sortino, Giovanna Vitaliti, Grete Francesca Privitera, Martino Ruggieri, Gaia Fusto, Xena Giada Pappalardo\",\"doi\":\"10.1007/s10048-023-00742-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Glucose transporter type 1 deficiency syndrome (GLUT-1DS) is characterized by alterations in glucose translocation through the blood-brain barrier (BBB) due to mutation involving the GLUT-1 transporter. The fundamental therapy is ketogenic diet (KD) that provide an alternative energetic substrate - ketone bodies that across the BBB via MCT-1 - for the brain. Symptoms are various and include intractable seizure, acquired microcephalia, abnormal ocular movement, movement disorder, and neurodevelopment delay secondary to an energetic crisis for persistent neuroglycopenia. KD is extremely effective in controlling epileptic seizures and has a positive impact on movement disorders and cognitive impairment. Cases of KD resistance are rare, and only a few of them are reported in the literature, all regarding seizure. Our study describes a peculiar case of GLUT-1DS due to a new deletion involving the first codon of SLC2A1 gene determining a loss of function with a resistance to KD admitted to hospital due to intractable episodes of dystonia. This patient presented a worsening of symptomatology at higher ketonemia values but without hyperketosis and showed a complete resolution of symptomatology while maintaining low ketonemia values. Our study proposes an in-silico genomic and proteomic analysis aimed at explaining the atypical response to KD exhibited by our patient. In this way, we propose a new clinical and research approach based on precision medicine and molecular modelling to be applied to patients with GLUT-1DS resistant to first-line treatment with ketogenic diet by in silico study of genetic and altered protein product.</p>\",\"PeriodicalId\":56106,\"journal\":{\"name\":\"Neurogenetics\",\"volume\":\" \",\"pages\":\"69-78\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2024-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurogenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10048-023-00742-8\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/8 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00742-8","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/8 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
GLUT-1DS resistant to ketogenic diet: from clinical feature to in silico analysis. An exemplificative case report with a literature review.
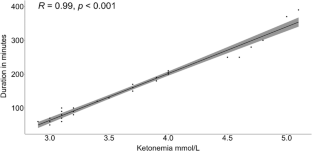
Glucose transporter type 1 deficiency syndrome (GLUT-1DS) is characterized by alterations in glucose translocation through the blood-brain barrier (BBB) due to mutation involving the GLUT-1 transporter. The fundamental therapy is ketogenic diet (KD) that provide an alternative energetic substrate - ketone bodies that across the BBB via MCT-1 - for the brain. Symptoms are various and include intractable seizure, acquired microcephalia, abnormal ocular movement, movement disorder, and neurodevelopment delay secondary to an energetic crisis for persistent neuroglycopenia. KD is extremely effective in controlling epileptic seizures and has a positive impact on movement disorders and cognitive impairment. Cases of KD resistance are rare, and only a few of them are reported in the literature, all regarding seizure. Our study describes a peculiar case of GLUT-1DS due to a new deletion involving the first codon of SLC2A1 gene determining a loss of function with a resistance to KD admitted to hospital due to intractable episodes of dystonia. This patient presented a worsening of symptomatology at higher ketonemia values but without hyperketosis and showed a complete resolution of symptomatology while maintaining low ketonemia values. Our study proposes an in-silico genomic and proteomic analysis aimed at explaining the atypical response to KD exhibited by our patient. In this way, we propose a new clinical and research approach based on precision medicine and molecular modelling to be applied to patients with GLUT-1DS resistant to first-line treatment with ketogenic diet by in silico study of genetic and altered protein product.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


