{"title":"用混合多态迭代玻尔兹曼反演法推导半晶聚合物的粗粒势垒","authors":"Omid Eghlidos, and , Jay Oswald*, ","doi":"10.1021/acs.jctc.3c00935","DOIUrl":null,"url":null,"abstract":"<p >In this article, we employ the multistate iterative Boltzmann inversion (MS-IBI) method to develop coarse-grained potentials capable of representing molecular structure in both the amorphous and crystalline phases of semicrystalline polymers with improved accuracy while allowing for tunable control over the dynamics governing the α-relaxation process. A unique feature of this method is that the potentials are blended using the product of the target structural distributions, for example, the radial density function, for each phase and a weighting factor. To demonstrate this approach, a family of potentials for polyethylene is developed where the weighting factor of the crystalline phase ranges is varied from zero, incorporating information only from the amorphous phase, to unity, where the model is trained from only the crystalline phase. The most accurate representation of structural distributions was obtained when the crystalline phases is weighted at 50%. However, we show that when the crystalline phase is weighted at 90%, the model more accurately represents dynamics of the α-relaxation process, with realistic predicted values of activation energy and diffusion rates, with relatively minor impact on accuracy in structure.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"19 24","pages":"9445–9456"},"PeriodicalIF":5.5000,"publicationDate":"2023-12-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Derived Coarse-Grained Potentials for Semicrystalline Polymers with a Blended Multistate Iterative Boltzmann Inversion Method\",\"authors\":\"Omid Eghlidos, and , Jay Oswald*, \",\"doi\":\"10.1021/acs.jctc.3c00935\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >In this article, we employ the multistate iterative Boltzmann inversion (MS-IBI) method to develop coarse-grained potentials capable of representing molecular structure in both the amorphous and crystalline phases of semicrystalline polymers with improved accuracy while allowing for tunable control over the dynamics governing the α-relaxation process. A unique feature of this method is that the potentials are blended using the product of the target structural distributions, for example, the radial density function, for each phase and a weighting factor. To demonstrate this approach, a family of potentials for polyethylene is developed where the weighting factor of the crystalline phase ranges is varied from zero, incorporating information only from the amorphous phase, to unity, where the model is trained from only the crystalline phase. The most accurate representation of structural distributions was obtained when the crystalline phases is weighted at 50%. However, we show that when the crystalline phase is weighted at 90%, the model more accurately represents dynamics of the α-relaxation process, with realistic predicted values of activation energy and diffusion rates, with relatively minor impact on accuracy in structure.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"19 24\",\"pages\":\"9445–9456\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2023-12-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.3c00935\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.3c00935","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Derived Coarse-Grained Potentials for Semicrystalline Polymers with a Blended Multistate Iterative Boltzmann Inversion Method
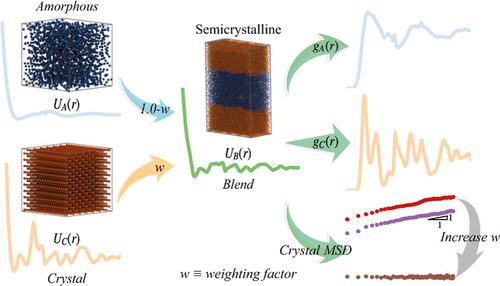
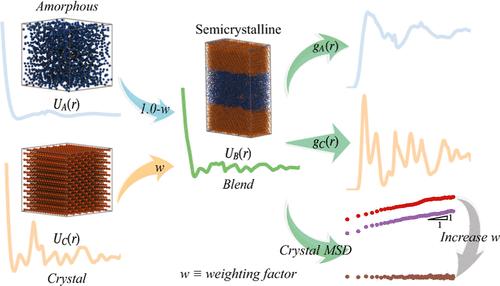
In this article, we employ the multistate iterative Boltzmann inversion (MS-IBI) method to develop coarse-grained potentials capable of representing molecular structure in both the amorphous and crystalline phases of semicrystalline polymers with improved accuracy while allowing for tunable control over the dynamics governing the α-relaxation process. A unique feature of this method is that the potentials are blended using the product of the target structural distributions, for example, the radial density function, for each phase and a weighting factor. To demonstrate this approach, a family of potentials for polyethylene is developed where the weighting factor of the crystalline phase ranges is varied from zero, incorporating information only from the amorphous phase, to unity, where the model is trained from only the crystalline phase. The most accurate representation of structural distributions was obtained when the crystalline phases is weighted at 50%. However, we show that when the crystalline phase is weighted at 90%, the model more accurately represents dynamics of the α-relaxation process, with realistic predicted values of activation energy and diffusion rates, with relatively minor impact on accuracy in structure.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


