{"title":"用于 mRNA 修饰全转录组定量的碱基分辨率测序方法","authors":"Li-Sheng Zhang, Qing Dai and Chuan He*, ","doi":"10.1021/acs.accounts.3c00532","DOIUrl":null,"url":null,"abstract":"<p >RNA molecules are not merely a combination of four bases of A, C, G, and U. Chemical modifications occur in almost all RNA species and play diverse roles in gene expression regulation. The abundant cellular RNAs, such as ribosomal RNA (rRNA) and transfer RNA (tRNA), are known to have the highest density of RNA modifications, which exert critical functions in rRNA and tRNA biogenesis, stability, and subsequent translation. In recent years, modifications on low-abundance RNA species in mammalian cells, such as messenger RNA (mRNA), regulatory noncoding RNA (ncRNA), and chromatin-associated RNA (caRNA), have been shown to contain multiple different chemical modifications with functional significance.</p><p >As the most abundant mRNA modification in mammals, <i>N</i><sup>6</sup>-methyladenosine (m<sup>6</sup>A) affects nearly every stage of mRNA processing and metabolism, with the antibody-based m<sup>6</sup>A-MeRIP-seq (methylated RNA immunoprecipitation sequencing) followed by high-throughput sequencing widely employed in mapping m<sup>6</sup>A distribution transcriptome-wide in diverse biological systems. In addition to m<sup>6</sup>A, other chemical modifications such as pseudouridine (Ψ), 2′-<i>O</i>-methylation (N<sub>m</sub>), 5-methylcytidine (m<sup>5</sup>C), internal <i>N</i><sup>7</sup>-methylguanosine (m<sup>7</sup>G), <i>N</i><sup>1</sup>-methyladenosine (m<sup>1</sup>A), <i>N</i><sup>4</sup>-acetylcytidine (ac<sup>4</sup>C), etc. also exist in polyA-tailed RNA in mammalian cells, requiring effective mapping approaches for whole-transcriptome profiling of these non-m<sup>6</sup>A mRNA modifications. Like m<sup>6</sup>A, the antibody-based enrichment followed by sequencing has been the primary method to study distributions of these modifications. Methods to more quantitatively map these modifications would dramatically improve our understanding of distributions and modification density of these chemical marks on RNA, thereby bettering informing functional implications. In this Account, aimed at both single-base resolution and modification fraction quantification, we summarize our recent advances in developing a series of chemistry- or biochemistry-based methods to quantitatively map RNA modifications, including m<sup>6</sup>A, Ψ, m<sup>5</sup>C, m<sup>1</sup>A, 2′-<i>O</i>-methylation (N<sub>m</sub>), and internal m<sup>7</sup>G, in mammalian mRNA at base resolution. These new methods, including m<sup>6</sup>A-SAC-seq, eTAM-seq, BID-seq, UBS-seq, DAMM-seq, m<sup>1</sup>A-quant-seq, Nm-Mut-seq, and m<sup>7</sup>G-quant-seq, promise to conduct base-resolution mapping of most major mRNA modifications with low RNA input and uncover dynamic changes in modification stoichiometry during biological and physiological processes, facilitating future investigations on these RNA modifications in regulating cellular gene expression and as potential biomarkers for clinical diagnosis and prognosis. These quantitative sequencing methods allow the mapping of most mRNA modifications with limited input sample requirements. The same modifications on diverse RNA species, such as caRNA, ncRNA, nuclear nascent RNA, mitochondrial RNA, cell-free RNA (cfRNA), etc., could be sequenced using the same methods.</p>","PeriodicalId":1,"journal":{"name":"Accounts of Chemical Research","volume":"57 1","pages":"47–58"},"PeriodicalIF":17.7000,"publicationDate":"2023-12-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.accounts.3c00532","citationCount":"0","resultStr":"{\"title\":\"Base-Resolution Sequencing Methods for Whole-Transcriptome Quantification of mRNA Modifications\",\"authors\":\"Li-Sheng Zhang, Qing Dai and Chuan He*, \",\"doi\":\"10.1021/acs.accounts.3c00532\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >RNA molecules are not merely a combination of four bases of A, C, G, and U. Chemical modifications occur in almost all RNA species and play diverse roles in gene expression regulation. The abundant cellular RNAs, such as ribosomal RNA (rRNA) and transfer RNA (tRNA), are known to have the highest density of RNA modifications, which exert critical functions in rRNA and tRNA biogenesis, stability, and subsequent translation. In recent years, modifications on low-abundance RNA species in mammalian cells, such as messenger RNA (mRNA), regulatory noncoding RNA (ncRNA), and chromatin-associated RNA (caRNA), have been shown to contain multiple different chemical modifications with functional significance.</p><p >As the most abundant mRNA modification in mammals, <i>N</i><sup>6</sup>-methyladenosine (m<sup>6</sup>A) affects nearly every stage of mRNA processing and metabolism, with the antibody-based m<sup>6</sup>A-MeRIP-seq (methylated RNA immunoprecipitation sequencing) followed by high-throughput sequencing widely employed in mapping m<sup>6</sup>A distribution transcriptome-wide in diverse biological systems. In addition to m<sup>6</sup>A, other chemical modifications such as pseudouridine (Ψ), 2′-<i>O</i>-methylation (N<sub>m</sub>), 5-methylcytidine (m<sup>5</sup>C), internal <i>N</i><sup>7</sup>-methylguanosine (m<sup>7</sup>G), <i>N</i><sup>1</sup>-methyladenosine (m<sup>1</sup>A), <i>N</i><sup>4</sup>-acetylcytidine (ac<sup>4</sup>C), etc. also exist in polyA-tailed RNA in mammalian cells, requiring effective mapping approaches for whole-transcriptome profiling of these non-m<sup>6</sup>A mRNA modifications. Like m<sup>6</sup>A, the antibody-based enrichment followed by sequencing has been the primary method to study distributions of these modifications. Methods to more quantitatively map these modifications would dramatically improve our understanding of distributions and modification density of these chemical marks on RNA, thereby bettering informing functional implications. In this Account, aimed at both single-base resolution and modification fraction quantification, we summarize our recent advances in developing a series of chemistry- or biochemistry-based methods to quantitatively map RNA modifications, including m<sup>6</sup>A, Ψ, m<sup>5</sup>C, m<sup>1</sup>A, 2′-<i>O</i>-methylation (N<sub>m</sub>), and internal m<sup>7</sup>G, in mammalian mRNA at base resolution. These new methods, including m<sup>6</sup>A-SAC-seq, eTAM-seq, BID-seq, UBS-seq, DAMM-seq, m<sup>1</sup>A-quant-seq, Nm-Mut-seq, and m<sup>7</sup>G-quant-seq, promise to conduct base-resolution mapping of most major mRNA modifications with low RNA input and uncover dynamic changes in modification stoichiometry during biological and physiological processes, facilitating future investigations on these RNA modifications in regulating cellular gene expression and as potential biomarkers for clinical diagnosis and prognosis. These quantitative sequencing methods allow the mapping of most mRNA modifications with limited input sample requirements. The same modifications on diverse RNA species, such as caRNA, ncRNA, nuclear nascent RNA, mitochondrial RNA, cell-free RNA (cfRNA), etc., could be sequenced using the same methods.</p>\",\"PeriodicalId\":1,\"journal\":{\"name\":\"Accounts of Chemical Research\",\"volume\":\"57 1\",\"pages\":\"47–58\"},\"PeriodicalIF\":17.7000,\"publicationDate\":\"2023-12-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/epdf/10.1021/acs.accounts.3c00532\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Accounts of Chemical Research\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.accounts.3c00532\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Accounts of Chemical Research","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.accounts.3c00532","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Base-Resolution Sequencing Methods for Whole-Transcriptome Quantification of mRNA Modifications
RNA molecules are not merely a combination of four bases of A, C, G, and U. Chemical modifications occur in almost all RNA species and play diverse roles in gene expression regulation. The abundant cellular RNAs, such as ribosomal RNA (rRNA) and transfer RNA (tRNA), are known to have the highest density of RNA modifications, which exert critical functions in rRNA and tRNA biogenesis, stability, and subsequent translation. In recent years, modifications on low-abundance RNA species in mammalian cells, such as messenger RNA (mRNA), regulatory noncoding RNA (ncRNA), and chromatin-associated RNA (caRNA), have been shown to contain multiple different chemical modifications with functional significance.
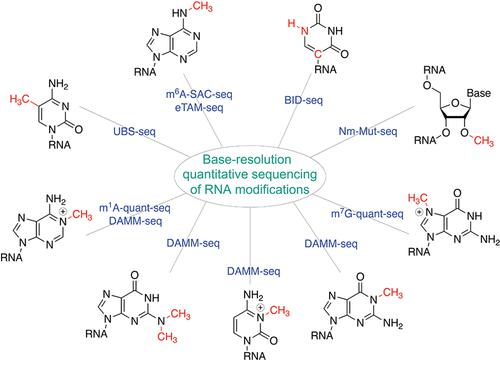
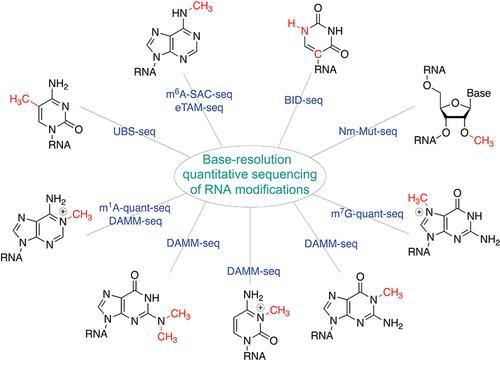
As the most abundant mRNA modification in mammals, N6-methyladenosine (m6A) affects nearly every stage of mRNA processing and metabolism, with the antibody-based m6A-MeRIP-seq (methylated RNA immunoprecipitation sequencing) followed by high-throughput sequencing widely employed in mapping m6A distribution transcriptome-wide in diverse biological systems. In addition to m6A, other chemical modifications such as pseudouridine (Ψ), 2′-O-methylation (Nm), 5-methylcytidine (m5C), internal N7-methylguanosine (m7G), N1-methyladenosine (m1A), N4-acetylcytidine (ac4C), etc. also exist in polyA-tailed RNA in mammalian cells, requiring effective mapping approaches for whole-transcriptome profiling of these non-m6A mRNA modifications. Like m6A, the antibody-based enrichment followed by sequencing has been the primary method to study distributions of these modifications. Methods to more quantitatively map these modifications would dramatically improve our understanding of distributions and modification density of these chemical marks on RNA, thereby bettering informing functional implications. In this Account, aimed at both single-base resolution and modification fraction quantification, we summarize our recent advances in developing a series of chemistry- or biochemistry-based methods to quantitatively map RNA modifications, including m6A, Ψ, m5C, m1A, 2′-O-methylation (Nm), and internal m7G, in mammalian mRNA at base resolution. These new methods, including m6A-SAC-seq, eTAM-seq, BID-seq, UBS-seq, DAMM-seq, m1A-quant-seq, Nm-Mut-seq, and m7G-quant-seq, promise to conduct base-resolution mapping of most major mRNA modifications with low RNA input and uncover dynamic changes in modification stoichiometry during biological and physiological processes, facilitating future investigations on these RNA modifications in regulating cellular gene expression and as potential biomarkers for clinical diagnosis and prognosis. These quantitative sequencing methods allow the mapping of most mRNA modifications with limited input sample requirements. The same modifications on diverse RNA species, such as caRNA, ncRNA, nuclear nascent RNA, mitochondrial RNA, cell-free RNA (cfRNA), etc., could be sequenced using the same methods.
期刊介绍:
Accounts of Chemical Research presents short, concise and critical articles offering easy-to-read overviews of basic research and applications in all areas of chemistry and biochemistry. These short reviews focus on research from the author’s own laboratory and are designed to teach the reader about a research project. In addition, Accounts of Chemical Research publishes commentaries that give an informed opinion on a current research problem. Special Issues online are devoted to a single topic of unusual activity and significance.
Accounts of Chemical Research replaces the traditional article abstract with an article "Conspectus." These entries synopsize the research affording the reader a closer look at the content and significance of an article. Through this provision of a more detailed description of the article contents, the Conspectus enhances the article's discoverability by search engines and the exposure for the research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


