Xijun Wang, Kaihang Shi, Anyang Peng and Randall Q. Snurr
{"title":"负载型氧化铜纳米团簇对甲烷活化的构效关系探讨","authors":"Xijun Wang, Kaihang Shi, Anyang Peng and Randall Q. Snurr","doi":"10.1039/D3EY00234A","DOIUrl":null,"url":null,"abstract":"<p >Supported metal oxide nanoclusters (MeO-NCs) have gained significant attention for their remarkable versatility in various energy and sustainability applications. Despite rapid advancements in atomic-scale synthesis and characterization techniques, the rational design of MeO-NCs with desired catalytic properties remains challenging. This challenge arises from the elusive and difficult-to-quantify structure-catalytic property relationships, particularly in the case of amorphous nanoclusters. Exploiting first-principles calculations at the density functional theory (DFT) level, we conducted a systematic investigation into the growth, geometries, and catalytic performance of a series of tetra-copper oxide nanoclusters (Cu<small><sub>4</sub></small>O-NCs) for methane activation. Focusing on the most representative geometries, we applied machine learning to extract two physically insightful descriptors involving the spin density, the p-band center of the oxygen site, and the d-band center of adjacent Cu sites. These descriptors enable us to predict free energy barriers associated with both the homolytic and heterolytic mechanisms of methane activation. This descriptor-driven approach enables rapid and intuitive prediction of the preferred reaction mechanism. Our findings lay a solid foundation for future advancements in catalysts based on amorphous nanoclusters and provide valuable insights into the mechanistic landscape of methane activation.</p>","PeriodicalId":72877,"journal":{"name":"EES catalysis","volume":" 1","pages":" 351-364"},"PeriodicalIF":0.0000,"publicationDate":"2023-11-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/ey/d3ey00234a?page=search","citationCount":"0","resultStr":"{\"title\":\"Probing the structure–property relationships of supported copper oxide nanoclusters for methane activation†\",\"authors\":\"Xijun Wang, Kaihang Shi, Anyang Peng and Randall Q. Snurr\",\"doi\":\"10.1039/D3EY00234A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Supported metal oxide nanoclusters (MeO-NCs) have gained significant attention for their remarkable versatility in various energy and sustainability applications. Despite rapid advancements in atomic-scale synthesis and characterization techniques, the rational design of MeO-NCs with desired catalytic properties remains challenging. This challenge arises from the elusive and difficult-to-quantify structure-catalytic property relationships, particularly in the case of amorphous nanoclusters. Exploiting first-principles calculations at the density functional theory (DFT) level, we conducted a systematic investigation into the growth, geometries, and catalytic performance of a series of tetra-copper oxide nanoclusters (Cu<small><sub>4</sub></small>O-NCs) for methane activation. Focusing on the most representative geometries, we applied machine learning to extract two physically insightful descriptors involving the spin density, the p-band center of the oxygen site, and the d-band center of adjacent Cu sites. These descriptors enable us to predict free energy barriers associated with both the homolytic and heterolytic mechanisms of methane activation. This descriptor-driven approach enables rapid and intuitive prediction of the preferred reaction mechanism. Our findings lay a solid foundation for future advancements in catalysts based on amorphous nanoclusters and provide valuable insights into the mechanistic landscape of methane activation.</p>\",\"PeriodicalId\":72877,\"journal\":{\"name\":\"EES catalysis\",\"volume\":\" 1\",\"pages\":\" 351-364\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-11-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2024/ey/d3ey00234a?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"EES catalysis\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/ey/d3ey00234a\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"EES catalysis","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/ey/d3ey00234a","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Probing the structure–property relationships of supported copper oxide nanoclusters for methane activation†
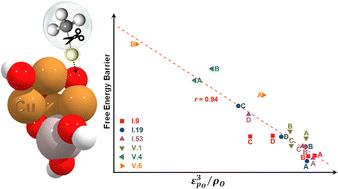
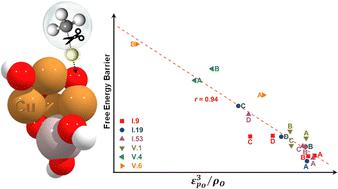
Supported metal oxide nanoclusters (MeO-NCs) have gained significant attention for their remarkable versatility in various energy and sustainability applications. Despite rapid advancements in atomic-scale synthesis and characterization techniques, the rational design of MeO-NCs with desired catalytic properties remains challenging. This challenge arises from the elusive and difficult-to-quantify structure-catalytic property relationships, particularly in the case of amorphous nanoclusters. Exploiting first-principles calculations at the density functional theory (DFT) level, we conducted a systematic investigation into the growth, geometries, and catalytic performance of a series of tetra-copper oxide nanoclusters (Cu4O-NCs) for methane activation. Focusing on the most representative geometries, we applied machine learning to extract two physically insightful descriptors involving the spin density, the p-band center of the oxygen site, and the d-band center of adjacent Cu sites. These descriptors enable us to predict free energy barriers associated with both the homolytic and heterolytic mechanisms of methane activation. This descriptor-driven approach enables rapid and intuitive prediction of the preferred reaction mechanism. Our findings lay a solid foundation for future advancements in catalysts based on amorphous nanoclusters and provide valuable insights into the mechanistic landscape of methane activation.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


