Katherine A English, Kate E Lines, Rajesh V Thakker
{"title":"原发性甲状旁腺功能亢进症遗传形式的遗传学。","authors":"Katherine A English, Kate E Lines, Rajesh V Thakker","doi":"10.1007/s42000-023-00508-9","DOIUrl":null,"url":null,"abstract":"<p><p>Primary hyperparathyroidism (PHPT), a relatively common disorder characterized by hypercalcemia with raised or inappropriately normal serum parathyroid hormone (PTH) concentrations, may occur as part of a hereditary syndromic disorder or as a non-syndromic disease. The associated syndromic disorders include multiple endocrine neoplasia types 1-5 (MEN1-5) and hyperparathyroidism with jaw tumor (HPT-JT) syndromes, and the non-syndromic forms include familial hypocalciuric hypercalcemia types 1-3 (FHH1-3), familial isolated hyperparathyroidism (FIHP), and neonatal severe hyperparathyroidism (NS-HPT). Such hereditary forms may occur in > 10% of patients with PHPT, and their recognition is important for implementation of gene-specific screening protocols and investigations for other associated tumors. Syndromic PHPT tends to be multifocal and multiglandular with most patients requiring parathyroidectomy with the aim of limiting end-organ damage associated with hypercalcemia, particularly osteoporosis, nephrolithiasis, and renal failure. Some patients with non-syndromic PHPT may have mutations of the MEN1 gene or the calcium-sensing receptor (CASR), whose loss of function mutations usually cause FHH1, a disorder associated with mild hypercalcemia and may follow a benign clinical course. Measurement of the urinary calcium-to-creatinine ratio clearance (UCCR) may help to distinguish patients with FHH from those with PHPT, as the majority of FHH patients have low urinary calcium excretion (UCCR < 0.01). Once genetic testing confirms a hereditary cause of PHPT, further genetic testing can be offered to the patients' relatives and subsequent screening can be carried out in these affected family members, which prevents inappropriate testing in normal individuals.</p>","PeriodicalId":50399,"journal":{"name":"Hormones-International Journal of Endocrinology and Metabolism","volume":" ","pages":"3-14"},"PeriodicalIF":2.4000,"publicationDate":"2024-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10847196/pdf/","citationCount":"0","resultStr":"{\"title\":\"Genetics of hereditary forms of primary hyperparathyroidism.\",\"authors\":\"Katherine A English, Kate E Lines, Rajesh V Thakker\",\"doi\":\"10.1007/s42000-023-00508-9\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Primary hyperparathyroidism (PHPT), a relatively common disorder characterized by hypercalcemia with raised or inappropriately normal serum parathyroid hormone (PTH) concentrations, may occur as part of a hereditary syndromic disorder or as a non-syndromic disease. The associated syndromic disorders include multiple endocrine neoplasia types 1-5 (MEN1-5) and hyperparathyroidism with jaw tumor (HPT-JT) syndromes, and the non-syndromic forms include familial hypocalciuric hypercalcemia types 1-3 (FHH1-3), familial isolated hyperparathyroidism (FIHP), and neonatal severe hyperparathyroidism (NS-HPT). Such hereditary forms may occur in > 10% of patients with PHPT, and their recognition is important for implementation of gene-specific screening protocols and investigations for other associated tumors. Syndromic PHPT tends to be multifocal and multiglandular with most patients requiring parathyroidectomy with the aim of limiting end-organ damage associated with hypercalcemia, particularly osteoporosis, nephrolithiasis, and renal failure. Some patients with non-syndromic PHPT may have mutations of the MEN1 gene or the calcium-sensing receptor (CASR), whose loss of function mutations usually cause FHH1, a disorder associated with mild hypercalcemia and may follow a benign clinical course. Measurement of the urinary calcium-to-creatinine ratio clearance (UCCR) may help to distinguish patients with FHH from those with PHPT, as the majority of FHH patients have low urinary calcium excretion (UCCR < 0.01). Once genetic testing confirms a hereditary cause of PHPT, further genetic testing can be offered to the patients' relatives and subsequent screening can be carried out in these affected family members, which prevents inappropriate testing in normal individuals.</p>\",\"PeriodicalId\":50399,\"journal\":{\"name\":\"Hormones-International Journal of Endocrinology and Metabolism\",\"volume\":\" \",\"pages\":\"3-14\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2024-03-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10847196/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Hormones-International Journal of Endocrinology and Metabolism\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s42000-023-00508-9\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/12/1 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hormones-International Journal of Endocrinology and Metabolism","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s42000-023-00508-9","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/12/1 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Genetics of hereditary forms of primary hyperparathyroidism.
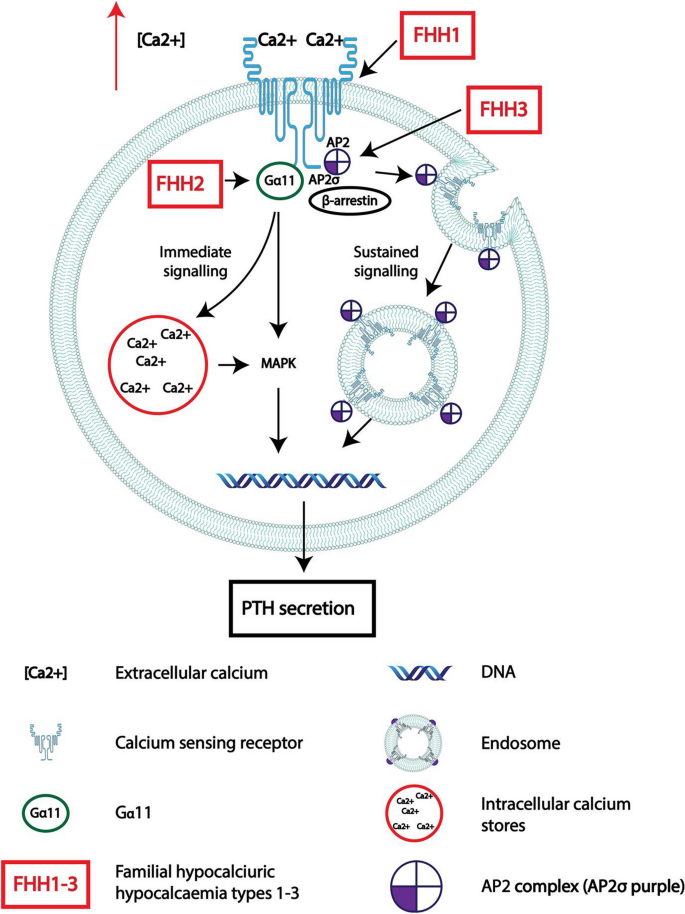
Primary hyperparathyroidism (PHPT), a relatively common disorder characterized by hypercalcemia with raised or inappropriately normal serum parathyroid hormone (PTH) concentrations, may occur as part of a hereditary syndromic disorder or as a non-syndromic disease. The associated syndromic disorders include multiple endocrine neoplasia types 1-5 (MEN1-5) and hyperparathyroidism with jaw tumor (HPT-JT) syndromes, and the non-syndromic forms include familial hypocalciuric hypercalcemia types 1-3 (FHH1-3), familial isolated hyperparathyroidism (FIHP), and neonatal severe hyperparathyroidism (NS-HPT). Such hereditary forms may occur in > 10% of patients with PHPT, and their recognition is important for implementation of gene-specific screening protocols and investigations for other associated tumors. Syndromic PHPT tends to be multifocal and multiglandular with most patients requiring parathyroidectomy with the aim of limiting end-organ damage associated with hypercalcemia, particularly osteoporosis, nephrolithiasis, and renal failure. Some patients with non-syndromic PHPT may have mutations of the MEN1 gene or the calcium-sensing receptor (CASR), whose loss of function mutations usually cause FHH1, a disorder associated with mild hypercalcemia and may follow a benign clinical course. Measurement of the urinary calcium-to-creatinine ratio clearance (UCCR) may help to distinguish patients with FHH from those with PHPT, as the majority of FHH patients have low urinary calcium excretion (UCCR < 0.01). Once genetic testing confirms a hereditary cause of PHPT, further genetic testing can be offered to the patients' relatives and subsequent screening can be carried out in these affected family members, which prevents inappropriate testing in normal individuals.
期刊介绍:
Hormones-International Journal of Endocrinology and Metabolism is an international journal published quarterly with an international editorial board aiming at providing a forum covering all fields of endocrinology and metabolic disorders such as disruption of glucose homeostasis (diabetes mellitus), impaired homeostasis of plasma lipids (dyslipidemia), the disorder of bone metabolism (osteoporosis), disturbances of endocrine function and reproductive capacity of women and men.
Hormones-International Journal of Endocrinology and Metabolism particularly encourages clinical, translational and basic science submissions in the areas of endocrine cancers, nutrition, obesity and metabolic disorders, quality of life of endocrine diseases, epidemiology of endocrine and metabolic disorders.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


