EXOSC8的错义变异导致外显子跳跃并扩大1C型桥小脑发育不全的表型谱。
IF 2.6
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
摘要
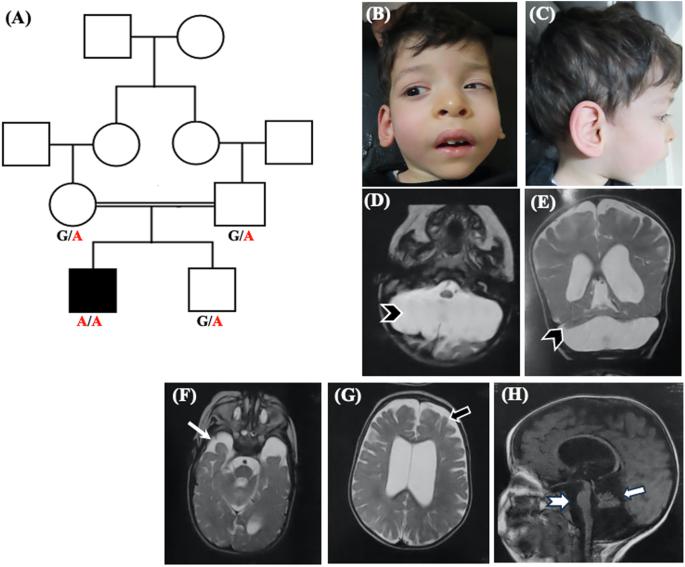
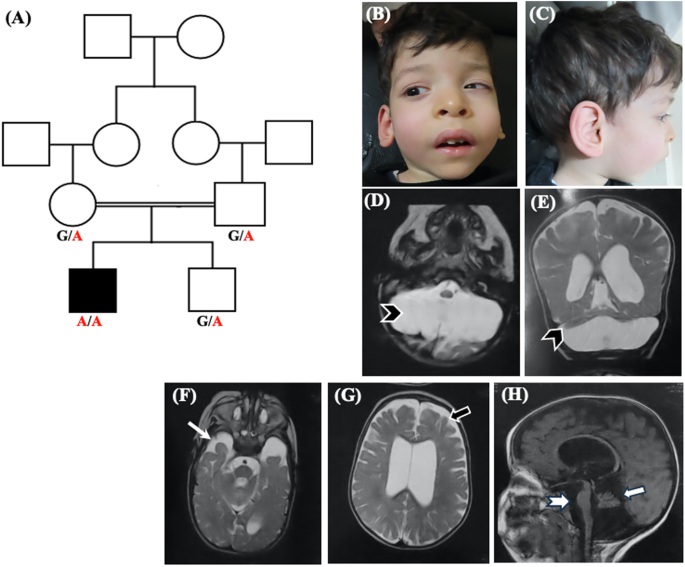
桥小脑发育不全(PCH)是一种罕见的影响桥桥和小脑的异质神经退行性疾病,目前分为17种类型(PCH1-PCH17)。PCH1与其他类型的脊髓运动神经元功能障碍有关。根据潜在的遗传病因,PCH1进一步分为6种不同的亚型(PCH1 A-F)。其中PCH 1C型是由EXOSC8基因的致病变异引起的,目前文献中只报道了4个家族。在这项研究中,我们报告了一名新的PCH1患者,他通过全外显子组测序证明在EXOSC8基因的剪接区含有一种新的纯合错义变体(c.238 G > a;p.Val80Ile)。对患者mRNA的研究证实,该变异导致基因外显子5的跳变和早期蛋白截断。我们的患者表现出PCH 1C型的主要临床表现包括精神运动迟缓、痉挛、脊髓肌萎缩和呼吸问题。然而,与大多数报告的病例不同,他没有出现听力或视力障碍,并表现出较长的生存期。此外,我们的患者有畸形相、眼球震颤、先天性内斜视和挛缩,这些在EXOSC8患者中很少出现。膈疝、侧脑室扩张、颞叶发育不良和脑干变薄是本例患者的新发现。本研究介绍了这种极为罕见的PCH的第五个家族,并扩大了相关的临床和脑成像结果。本文章由计算机程序翻译,如有差异,请以英文原文为准。
A missense variant in EXOSC8 causes exon skipping and expands the phenotypic spectrum of pontocerebellar hypoplasia type 1C
Pontocerebellar hypoplasia (PCH) is a rare heterogeneous neurodegenerative disorder affecting the pons and cerebellum and is currently classified into 17 types (PCH1-PCH17). PCH1 is distinguishable from other types by the association of spinal motor neuron dysfunction. Based on the underlying genetic etiology, PCH1 is further classified into 6 different subtypes (PCH1 A-F). Of them, PCH type 1C is caused by pathogenic variants in EXOSC8 gene and so far, only four families have been described in the literature. In this study, we report a new patient with PCH1 who proved by whole-exome sequencing to harbor a novel homozygous missense variant in the splice region of EXOSC8 gene (c.238 G > A; p.Val80Ile). Studying mRNA of the patient confirmed that this variant results in skipping of exon 5 of the gene and early protein truncation. Our patient presented with the main clinical findings of PCH type 1C including psychomotor retardation, spasticity, spinal muscle atrophy, and respiratory problems. However, unlike most of the reported cases, he did not develop hearing or visual impairment and displayed a longer survival. In addition, our patient had dysmorphic facies, nystagmus, congenital esotropia and contractures which were infrequently described in patients with EXOSC8. Diaphragmatic hernia, dilated lateral ventricles, hypoplastic temporal lobes, and thinning of the brain stem were additional new findings noted in our patient. This study presents the fifth family with this extremely rare type of PCH and expands the associated clinical and brain imaging findings.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


