Bijaya B. Karki, Dipta B. Ghosh, Jianwei Wang, Shun-ichiro Karato
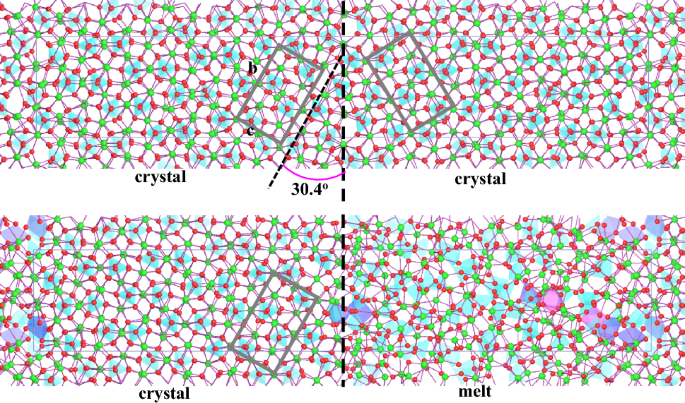
{"title":"高压下Mg2SiO4晶体-熔体界面:来自第一性原理模拟的结构和能量学见解","authors":"Bijaya B. Karki, Dipta B. Ghosh, Jianwei Wang, Shun-ichiro Karato","doi":"10.1007/s00269-023-01256-3","DOIUrl":null,"url":null,"abstract":"<div><p>The interplay between crystal–melt and grain boundary interfaces in partially melted polycrystalline aggregates controls many physical properties of mantle rocks. To understand this process at the fundamental level requires improved knowledge about the interfacial structures and energetics. Here, we report the results of first-principles molecular dynamics simulations of two grain boundaries of (0<i>l</i>1)/[100] type for tilt angles of 30.4° and 49.6° and the corresponding solid–liquid interfaces in Mg<sub>2</sub>SiO<sub>4</sub> forsterite at the conditions of the upper mantle. Our analysis of the simulated position time series shows that structural distortions at the solid–liquid interfacial region are stronger than intergranular interfacial distortions. The calculated formation enthalpy of the solid–solid interfaces increases nearly linearly from 1.0 to 1.4 J/m<sup>2</sup> for the 30.4° tilt and from 0.8 to 1.0 J/m<sup>2</sup> for the 49.6° tilt with pressure from 0 to 16 GPa at 1500 K, being consistent with the experimental data. The solid–liquid interfacial enthalpy takes comparable values in the range 0.9 to 1.5 J/m<sup>2</sup> over similar pressure interval. The dihedral angle of the forsterite–melt system estimated using these interfacial enthalpies takes values in the range of 67° to 146°, showing a decreasing trend with pressure. The predicted dihedral angle is found to be generally larger than the measured data for silicate systems, probably caused by compositional differences between the simulation and the measurements.</p></div>","PeriodicalId":20132,"journal":{"name":"Physics and Chemistry of Minerals","volume":"50 4","pages":""},"PeriodicalIF":1.6000,"publicationDate":"2023-10-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s00269-023-01256-3.pdf","citationCount":"0","resultStr":"{\"title\":\"Crystal–melt interfaces in Mg2SiO4 at high pressure: structural and energetics insights from first-principles simulations\",\"authors\":\"Bijaya B. Karki, Dipta B. Ghosh, Jianwei Wang, Shun-ichiro Karato\",\"doi\":\"10.1007/s00269-023-01256-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The interplay between crystal–melt and grain boundary interfaces in partially melted polycrystalline aggregates controls many physical properties of mantle rocks. To understand this process at the fundamental level requires improved knowledge about the interfacial structures and energetics. Here, we report the results of first-principles molecular dynamics simulations of two grain boundaries of (0<i>l</i>1)/[100] type for tilt angles of 30.4° and 49.6° and the corresponding solid–liquid interfaces in Mg<sub>2</sub>SiO<sub>4</sub> forsterite at the conditions of the upper mantle. Our analysis of the simulated position time series shows that structural distortions at the solid–liquid interfacial region are stronger than intergranular interfacial distortions. The calculated formation enthalpy of the solid–solid interfaces increases nearly linearly from 1.0 to 1.4 J/m<sup>2</sup> for the 30.4° tilt and from 0.8 to 1.0 J/m<sup>2</sup> for the 49.6° tilt with pressure from 0 to 16 GPa at 1500 K, being consistent with the experimental data. The solid–liquid interfacial enthalpy takes comparable values in the range 0.9 to 1.5 J/m<sup>2</sup> over similar pressure interval. The dihedral angle of the forsterite–melt system estimated using these interfacial enthalpies takes values in the range of 67° to 146°, showing a decreasing trend with pressure. The predicted dihedral angle is found to be generally larger than the measured data for silicate systems, probably caused by compositional differences between the simulation and the measurements.</p></div>\",\"PeriodicalId\":20132,\"journal\":{\"name\":\"Physics and Chemistry of Minerals\",\"volume\":\"50 4\",\"pages\":\"\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2023-10-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://link.springer.com/content/pdf/10.1007/s00269-023-01256-3.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physics and Chemistry of Minerals\",\"FirstCategoryId\":\"89\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00269-023-01256-3\",\"RegionNum\":4,\"RegionCategory\":\"地球科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"MATERIALS SCIENCE, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physics and Chemistry of Minerals","FirstCategoryId":"89","ListUrlMain":"https://link.springer.com/article/10.1007/s00269-023-01256-3","RegionNum":4,"RegionCategory":"地球科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
Crystal–melt interfaces in Mg2SiO4 at high pressure: structural and energetics insights from first-principles simulations
The interplay between crystal–melt and grain boundary interfaces in partially melted polycrystalline aggregates controls many physical properties of mantle rocks. To understand this process at the fundamental level requires improved knowledge about the interfacial structures and energetics. Here, we report the results of first-principles molecular dynamics simulations of two grain boundaries of (0l1)/[100] type for tilt angles of 30.4° and 49.6° and the corresponding solid–liquid interfaces in Mg2SiO4 forsterite at the conditions of the upper mantle. Our analysis of the simulated position time series shows that structural distortions at the solid–liquid interfacial region are stronger than intergranular interfacial distortions. The calculated formation enthalpy of the solid–solid interfaces increases nearly linearly from 1.0 to 1.4 J/m2 for the 30.4° tilt and from 0.8 to 1.0 J/m2 for the 49.6° tilt with pressure from 0 to 16 GPa at 1500 K, being consistent with the experimental data. The solid–liquid interfacial enthalpy takes comparable values in the range 0.9 to 1.5 J/m2 over similar pressure interval. The dihedral angle of the forsterite–melt system estimated using these interfacial enthalpies takes values in the range of 67° to 146°, showing a decreasing trend with pressure. The predicted dihedral angle is found to be generally larger than the measured data for silicate systems, probably caused by compositional differences between the simulation and the measurements.
期刊介绍:
Physics and Chemistry of Minerals is an international journal devoted to publishing articles and short communications of physical or chemical studies on minerals or solids related to minerals. The aim of the journal is to support competent interdisciplinary work in mineralogy and physics or chemistry. Particular emphasis is placed on applications of modern techniques or new theories and models to interpret atomic structures and physical or chemical properties of minerals. Some subjects of interest are:
-Relationships between atomic structure and crystalline state (structures of various states, crystal energies, crystal growth, thermodynamic studies, phase transformations, solid solution, exsolution phenomena, etc.)
-General solid state spectroscopy (ultraviolet, visible, infrared, Raman, ESCA, luminescence, X-ray, electron paramagnetic resonance, nuclear magnetic resonance, gamma ray resonance, etc.)
-Experimental and theoretical analysis of chemical bonding in minerals (application of crystal field, molecular orbital, band theories, etc.)
-Physical properties (magnetic, mechanical, electric, optical, thermodynamic, etc.)
-Relations between thermal expansion, compressibility, elastic constants, and fundamental properties of atomic structure, particularly as applied to geophysical problems
-Electron microscopy in support of physical and chemical studies
-Computational methods in the study of the structure and properties of minerals
-Mineral surfaces (experimental methods, structure and properties)

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


