{"title":"报道两个伊朗囊性纤维化家族的两个新突变,分子和生物信息学分析","authors":"Amin Hosseini Nami, Mahboubeh Kabiri, Sirous Zeinali","doi":"10.52547/ibj. 3713","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Cystic fibrosis (CF) is the most common heredity disease among the Caucasian population. More than 350 known pathogenic variations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene (NM_000492.4) cause CF. Herein, we report the outcome of our investigation in two unrelated Iranian families with CF patients.</p><p><strong>Methods: </strong>We conducted phenotypic examination, segregation, linkage analysis, and CFTR gene sequencing to define causative mutations.</p><p><strong>Results: </strong>We found two novel mutations in the present study. The first one was a deletion causing frameshift, c.299delT p.(Leu100Profs*7), and the second one was a missense mutation, c.1857G>T, at nucleotide binding domain 1 of the CFTR protein. Haplotype segregation data supported our new mutation findings.</p><p><strong>Conclusion: </strong>Findings of this study expand the spectrum of CFTR pathogenic variations and can improve prenatal diagnosis and genetic counseling for CF.</p>","PeriodicalId":14500,"journal":{"name":"Iranian Biomedical Journal","volume":"26 5","pages":"398-405"},"PeriodicalIF":0.0000,"publicationDate":"2022-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9763878/pdf/","citationCount":"0","resultStr":"{\"title\":\"Reporting Two Novel Mutations in Two Iranian Families with Cystic Fibrosis, Molecular and Bioinformatic Analysis\",\"authors\":\"Amin Hosseini Nami, Mahboubeh Kabiri, Sirous Zeinali\",\"doi\":\"10.52547/ibj. 3713\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Cystic fibrosis (CF) is the most common heredity disease among the Caucasian population. More than 350 known pathogenic variations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene (NM_000492.4) cause CF. Herein, we report the outcome of our investigation in two unrelated Iranian families with CF patients.</p><p><strong>Methods: </strong>We conducted phenotypic examination, segregation, linkage analysis, and CFTR gene sequencing to define causative mutations.</p><p><strong>Results: </strong>We found two novel mutations in the present study. The first one was a deletion causing frameshift, c.299delT p.(Leu100Profs*7), and the second one was a missense mutation, c.1857G>T, at nucleotide binding domain 1 of the CFTR protein. Haplotype segregation data supported our new mutation findings.</p><p><strong>Conclusion: </strong>Findings of this study expand the spectrum of CFTR pathogenic variations and can improve prenatal diagnosis and genetic counseling for CF.</p>\",\"PeriodicalId\":14500,\"journal\":{\"name\":\"Iranian Biomedical Journal\",\"volume\":\"26 5\",\"pages\":\"398-405\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9763878/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Iranian Biomedical Journal\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.52547/ibj. 3713\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Iranian Biomedical Journal","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.52547/ibj. 3713","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Reporting Two Novel Mutations in Two Iranian Families with Cystic Fibrosis, Molecular and Bioinformatic Analysis
Background: Cystic fibrosis (CF) is the most common heredity disease among the Caucasian population. More than 350 known pathogenic variations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene (NM_000492.4) cause CF. Herein, we report the outcome of our investigation in two unrelated Iranian families with CF patients.
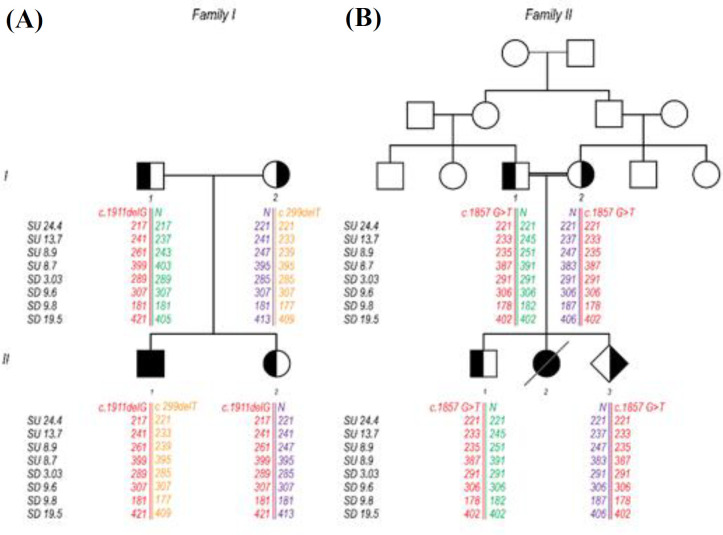
Methods: We conducted phenotypic examination, segregation, linkage analysis, and CFTR gene sequencing to define causative mutations.
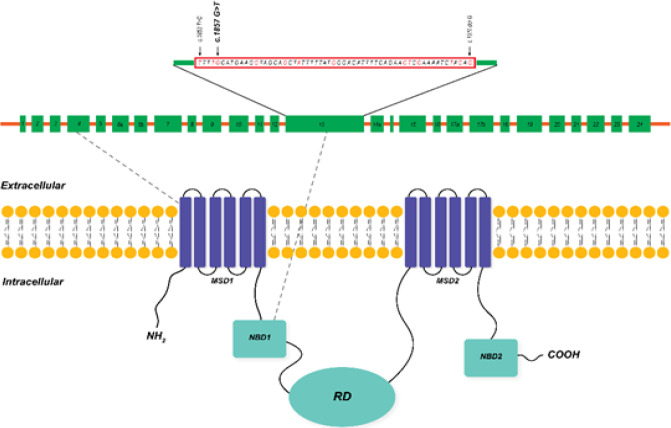
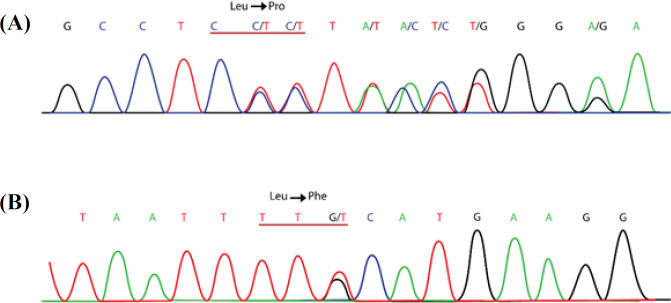
Results: We found two novel mutations in the present study. The first one was a deletion causing frameshift, c.299delT p.(Leu100Profs*7), and the second one was a missense mutation, c.1857G>T, at nucleotide binding domain 1 of the CFTR protein. Haplotype segregation data supported our new mutation findings.
Conclusion: Findings of this study expand the spectrum of CFTR pathogenic variations and can improve prenatal diagnosis and genetic counseling for CF.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


