Andres Nascimento, Christine C. Bruels, Sandra Donkervoort, A. Reghan Foley, Anna Codina, Jose C. Milisenda, Elicia A. Estrella, Chengcheng Li, Jordi Pijuan, Isabelle Draper, Ying Hu, Seth A. Stafki, Lynn S. Pais, Vijay S. Ganesh, Anne O’Donnell-Luria, Safoora B. Syeda, Laura Carrera-García, Jessica Expósito-Escudero, Delia Yubero, Loreto Martorell, Iago Pinal-Fernandez, Hart G. W. Lidov, Andrew L. Mammen, Josep M. Grau-Junyent, Carlos Ortez, Francesc Palau, Partha S. Ghosh, Basil T. Darras, Cristina Jou, Louis M. Kunkel, Janet Hoenicka, Carsten G. Bönnemann, Peter B. Kang, Daniel Natera-de Benito
{"title":"DTNA变异导致轻度显性遗传性肌营养不良","authors":"Andres Nascimento, Christine C. Bruels, Sandra Donkervoort, A. Reghan Foley, Anna Codina, Jose C. Milisenda, Elicia A. Estrella, Chengcheng Li, Jordi Pijuan, Isabelle Draper, Ying Hu, Seth A. Stafki, Lynn S. Pais, Vijay S. Ganesh, Anne O’Donnell-Luria, Safoora B. Syeda, Laura Carrera-García, Jessica Expósito-Escudero, Delia Yubero, Loreto Martorell, Iago Pinal-Fernandez, Hart G. W. Lidov, Andrew L. Mammen, Josep M. Grau-Junyent, Carlos Ortez, Francesc Palau, Partha S. Ghosh, Basil T. Darras, Cristina Jou, Louis M. Kunkel, Janet Hoenicka, Carsten G. Bönnemann, Peter B. Kang, Daniel Natera-de Benito","doi":"10.1007/s00401-023-02551-7","DOIUrl":null,"url":null,"abstract":"<div><p><i>DTNA</i> encodes α-dystrobrevin, a component of the macromolecular dystrophin–glycoprotein complex (DGC) that binds to dystrophin/utrophin and α-syntrophin. Mice lacking α-dystrobrevin have a muscular dystrophy phenotype, but variants in <i>DTNA</i> have not previously been associated with human skeletal muscle disease. We present 12 individuals from four unrelated families with two different monoallelic <i>DTNA</i> variants affecting the coiled-coil domain of α-dystrobrevin. The five affected individuals from family A harbor a c.1585G > A; p.Glu529Lys variant, while the recurrent c.1567_1587del; p.Gln523_Glu529del <i>DTNA</i> variant was identified in the other three families (family B: four affected individuals, family C: one affected individual, and family D: two affected individuals). Myalgia and exercise intolerance, with variable ages of onset, were reported in 10 of 12 affected individuals. Proximal lower limb weakness with onset in the first decade of life was noted in three individuals. Persistent elevations of serum creatine kinase (CK) levels were detected in 11 of 12 affected individuals, 1 of whom had an episode of rhabdomyolysis at 20 years of age. Autism spectrum disorder or learning disabilities were reported in four individuals with the c.1567_1587 deletion. Muscle biopsies in eight affected individuals showed mixed myopathic and dystrophic findings, characterized by fiber size variability, internalized nuclei, and slightly increased extracellular connective tissue and inflammation. Immunofluorescence analysis of biopsies from five affected individuals showed reduced α-dystrobrevin immunoreactivity and variably reduced immunoreactivity of other DGC proteins: dystrophin, α, β, δ and γ-sarcoglycans, and α and β-dystroglycans. The <i>DTNA</i> deletion disrupted an interaction between α-dystrobrevin and syntrophin. Specific variants in the coiled-coil domain of <i>DTNA</i> cause skeletal muscle disease with variable penetrance. Affected individuals show a spectrum of clinical manifestations, with severity ranging from hyperCKemia, myalgias, and exercise intolerance to childhood-onset proximal muscle weakness. Our findings expand the molecular etiologies of both muscular dystrophy and paucisymptomatic hyperCKemia, to now include monoallelic <i>DTNA</i> variants as a novel cause of skeletal muscle disease in humans.</p></div>","PeriodicalId":7012,"journal":{"name":"Acta Neuropathologica","volume":"145 4","pages":"479 - 496"},"PeriodicalIF":9.3000,"publicationDate":"2023-02-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Variants in DTNA cause a mild, dominantly inherited muscular dystrophy\",\"authors\":\"Andres Nascimento, Christine C. Bruels, Sandra Donkervoort, A. Reghan Foley, Anna Codina, Jose C. Milisenda, Elicia A. Estrella, Chengcheng Li, Jordi Pijuan, Isabelle Draper, Ying Hu, Seth A. Stafki, Lynn S. Pais, Vijay S. Ganesh, Anne O’Donnell-Luria, Safoora B. Syeda, Laura Carrera-García, Jessica Expósito-Escudero, Delia Yubero, Loreto Martorell, Iago Pinal-Fernandez, Hart G. W. Lidov, Andrew L. Mammen, Josep M. Grau-Junyent, Carlos Ortez, Francesc Palau, Partha S. Ghosh, Basil T. Darras, Cristina Jou, Louis M. Kunkel, Janet Hoenicka, Carsten G. Bönnemann, Peter B. Kang, Daniel Natera-de Benito\",\"doi\":\"10.1007/s00401-023-02551-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p><i>DTNA</i> encodes α-dystrobrevin, a component of the macromolecular dystrophin–glycoprotein complex (DGC) that binds to dystrophin/utrophin and α-syntrophin. Mice lacking α-dystrobrevin have a muscular dystrophy phenotype, but variants in <i>DTNA</i> have not previously been associated with human skeletal muscle disease. We present 12 individuals from four unrelated families with two different monoallelic <i>DTNA</i> variants affecting the coiled-coil domain of α-dystrobrevin. The five affected individuals from family A harbor a c.1585G > A; p.Glu529Lys variant, while the recurrent c.1567_1587del; p.Gln523_Glu529del <i>DTNA</i> variant was identified in the other three families (family B: four affected individuals, family C: one affected individual, and family D: two affected individuals). Myalgia and exercise intolerance, with variable ages of onset, were reported in 10 of 12 affected individuals. Proximal lower limb weakness with onset in the first decade of life was noted in three individuals. Persistent elevations of serum creatine kinase (CK) levels were detected in 11 of 12 affected individuals, 1 of whom had an episode of rhabdomyolysis at 20 years of age. Autism spectrum disorder or learning disabilities were reported in four individuals with the c.1567_1587 deletion. Muscle biopsies in eight affected individuals showed mixed myopathic and dystrophic findings, characterized by fiber size variability, internalized nuclei, and slightly increased extracellular connective tissue and inflammation. Immunofluorescence analysis of biopsies from five affected individuals showed reduced α-dystrobrevin immunoreactivity and variably reduced immunoreactivity of other DGC proteins: dystrophin, α, β, δ and γ-sarcoglycans, and α and β-dystroglycans. The <i>DTNA</i> deletion disrupted an interaction between α-dystrobrevin and syntrophin. Specific variants in the coiled-coil domain of <i>DTNA</i> cause skeletal muscle disease with variable penetrance. Affected individuals show a spectrum of clinical manifestations, with severity ranging from hyperCKemia, myalgias, and exercise intolerance to childhood-onset proximal muscle weakness. Our findings expand the molecular etiologies of both muscular dystrophy and paucisymptomatic hyperCKemia, to now include monoallelic <i>DTNA</i> variants as a novel cause of skeletal muscle disease in humans.</p></div>\",\"PeriodicalId\":7012,\"journal\":{\"name\":\"Acta Neuropathologica\",\"volume\":\"145 4\",\"pages\":\"479 - 496\"},\"PeriodicalIF\":9.3000,\"publicationDate\":\"2023-02-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta Neuropathologica\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00401-023-02551-7\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Neuropathologica","FirstCategoryId":"3","ListUrlMain":"https://link.springer.com/article/10.1007/s00401-023-02551-7","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Variants in DTNA cause a mild, dominantly inherited muscular dystrophy
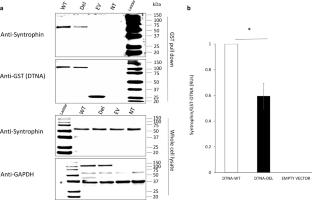
DTNA encodes α-dystrobrevin, a component of the macromolecular dystrophin–glycoprotein complex (DGC) that binds to dystrophin/utrophin and α-syntrophin. Mice lacking α-dystrobrevin have a muscular dystrophy phenotype, but variants in DTNA have not previously been associated with human skeletal muscle disease. We present 12 individuals from four unrelated families with two different monoallelic DTNA variants affecting the coiled-coil domain of α-dystrobrevin. The five affected individuals from family A harbor a c.1585G > A; p.Glu529Lys variant, while the recurrent c.1567_1587del; p.Gln523_Glu529del DTNA variant was identified in the other three families (family B: four affected individuals, family C: one affected individual, and family D: two affected individuals). Myalgia and exercise intolerance, with variable ages of onset, were reported in 10 of 12 affected individuals. Proximal lower limb weakness with onset in the first decade of life was noted in three individuals. Persistent elevations of serum creatine kinase (CK) levels were detected in 11 of 12 affected individuals, 1 of whom had an episode of rhabdomyolysis at 20 years of age. Autism spectrum disorder or learning disabilities were reported in four individuals with the c.1567_1587 deletion. Muscle biopsies in eight affected individuals showed mixed myopathic and dystrophic findings, characterized by fiber size variability, internalized nuclei, and slightly increased extracellular connective tissue and inflammation. Immunofluorescence analysis of biopsies from five affected individuals showed reduced α-dystrobrevin immunoreactivity and variably reduced immunoreactivity of other DGC proteins: dystrophin, α, β, δ and γ-sarcoglycans, and α and β-dystroglycans. The DTNA deletion disrupted an interaction between α-dystrobrevin and syntrophin. Specific variants in the coiled-coil domain of DTNA cause skeletal muscle disease with variable penetrance. Affected individuals show a spectrum of clinical manifestations, with severity ranging from hyperCKemia, myalgias, and exercise intolerance to childhood-onset proximal muscle weakness. Our findings expand the molecular etiologies of both muscular dystrophy and paucisymptomatic hyperCKemia, to now include monoallelic DTNA variants as a novel cause of skeletal muscle disease in humans.
期刊介绍:
Acta Neuropathologica publishes top-quality papers on the pathology of neurological diseases and experimental studies on molecular and cellular mechanisms using in vitro and in vivo models, ideally validated by analysis of human tissues. The journal accepts Original Papers, Review Articles, Case Reports, and Scientific Correspondence (Letters). Manuscripts must adhere to ethical standards, including review by appropriate ethics committees for human studies and compliance with principles of laboratory animal care for animal experiments. Failure to comply may result in rejection of the manuscript, and authors are responsible for ensuring accuracy and adherence to these requirements.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


