Serena Galosi MD, PhD, Maria Novelli MD, Martina Di Rocco PhD, Elisabetta Flex PhD, Elena Messina PhD, Luca Pollini MD, Elena Parrini PhD, Francesco Pisani MD, Renzo Guerrini MD, FRCP, Vincenzo Leuzzi MD, Simone Martinelli PhD
{"title":"GNAO1单倍体缺陷:GNAO1表型谱系中较温和的一端。","authors":"Serena Galosi MD, PhD, Maria Novelli MD, Martina Di Rocco PhD, Elisabetta Flex PhD, Elena Messina PhD, Luca Pollini MD, Elena Parrini PhD, Francesco Pisani MD, Renzo Guerrini MD, FRCP, Vincenzo Leuzzi MD, Simone Martinelli PhD","doi":"10.1002/mds.29585","DOIUrl":null,"url":null,"abstract":"<p>\n <i>GNAO1</i> variants are typically associated with severe, early-onset movement disorders (MDs) with life-threatening and drug-resistant paroxysmal exacerbations, neurodevelopmental disorders, and epilepsy. Recently, the phenotypic spectrum has broadened to include milder phenotypes with late-onset dystonia, minor cognitive impairment, and other neurological signs, including parkinsonism and myoclonus. <i>GNAO1</i> haploinsufficiency has been evoked as a putative mechanism underlying milder clinical presentations.<span><sup>1, 2</sup></span> To date, however, the functional consequences of this class of variants have not yet been evaluated.</p><p>We report on an 8-year-old boy with subtle neurological signs, including generalized tonic–clonic seizures during fever, mild language impairment, dystonic postures of lower limbs during walking, and occasional tongue dyskinetic movements (see Data S1 for more details and Video 1). A next generation sequencing–based epilepsy panel revealed a de novo NM_020988.3:c.163_164del variant in <i>GNAO1</i>. No additional candidate variants were identified. Reverse transcription polymerase chain reaction showed an approximately 50% decrease in the expression of the endogenous <i>GNAO1</i> gene in cells from the affected child compared with cells from the unaffected father (Figs. 1A and S2), suggesting nonsense-mediated mRNA decay (NMD). If translated, the c.163_164delAT allele was predicted to generate a truncated protein (p.Ile55Hisfs*3). As expected, Western blotting performed in transiently transfected HEK293T cells revealed the lack of the truncated form of Gαo (Fig. 1B), which was not restored by MG132 or bafilomycin treatments, inhibitors of the ubiquitin/proteasome and autophagy pathways, respectively. These findings demonstrate that the c.163_164delAT transcript undergoes NMD, leading to <i>GNAO1</i> haploinsufficiency.</p><p>Genotype/phenotype correlations in <i>GNAO1</i> encephalopathy are still far from being elucidated. Recent studies suggest that pathogenic variants have a loss-of-function effect on Gαo-mediated signaling,<span><sup>3-7</sup></span> but the consequences on G-beta-gamma subunit (Gβγ) signaling that regulates cyclic adenosine monophosphate production remain unclear. Emerging data show that haploinsufficiency is associated with milder clinical features and later onset than missense changes underlying developmental and epileptic encephalopathy type 17 (Mendelian inheritance in man [MIM]#615473) or neurodevelopmental disorder with involuntary movements (MIM#617493). This finding has important implications. First, given the different phenotypic output, variants associated with the canonical form of <i>GNAO1</i> encephalopathy cannot have a simple loss-of-function effect; rather, they behave as dominant-negative alleles or alter Gα/Gβγ association, as recently shown for a subset of changes.<span><sup>3-7</sup></span> Second, the phenotype associated with <i>GNAO1</i> haploinsufficiency is likely attributed to increased levels of free Gβγ in the brain, which, in turn, could lead to increased receptor-independent Gβγ signaling in neurons. Finally, the association of <i>GNAO1</i> haploinsufficiency with a subtle but distinctive phenotype may help to design a proper gene therapy strategy. Allele-specific silencing by antisense oligonucleotides or short-interfering RNAs is unlikely to be a reasonable approach because 50% of the gene dosage is not neutral and single nucleotide substitutions hardly confer a complete discrimination for allele-specific targeting. In contrast, AAV-mediated gene supplementation coupled with silencing of the mutant allele is expected to effectively alleviate the disease phenotype.</p><p>Our findings also indicate that <i>GNAO1</i> variants may be more frequent than previously estimated and encourage testing for this gene in patients with mild neurological signs featuring epilepsy and/or MDs without a definite diagnosis. The progression into more severe phenotypes, and possible neurological deterioration induced by triggering events, typical for this condition, deserve a careful and prolonged clinical follow-up.</p><p>(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.</p><p>S.G.: 1A, 1B, 2B, 2C, 3A, 3B</p><p>M.N.: 1A, 1B, 3A, 3B</p><p>M.R.: 2B, 2C</p><p>E.F.: 3B</p><p>E.M.: 3B</p><p>L.P.: 3B</p><p>E.P.: 2B, 2C</p><p>F.P.: 3B</p><p>R.G.: 3B</p><p>V.L.: 1A, 1B, 3A, 3B</p><p>S.M.: 1A, 1B, 2B, 2C, 3A, 3B</p><p>None.</p>","PeriodicalId":213,"journal":{"name":"Movement Disorders","volume":"38 12","pages":"2313-2314"},"PeriodicalIF":7.4000,"publicationDate":"2023-08-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://movementdisorders.onlinelibrary.wiley.com/doi/epdf/10.1002/mds.29585","citationCount":"0","resultStr":"{\"title\":\"GNAO1 Haploinsufficiency: The Milder End of the GNAO1 Phenotypic Spectrum\",\"authors\":\"Serena Galosi MD, PhD, Maria Novelli MD, Martina Di Rocco PhD, Elisabetta Flex PhD, Elena Messina PhD, Luca Pollini MD, Elena Parrini PhD, Francesco Pisani MD, Renzo Guerrini MD, FRCP, Vincenzo Leuzzi MD, Simone Martinelli PhD\",\"doi\":\"10.1002/mds.29585\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>\\n <i>GNAO1</i> variants are typically associated with severe, early-onset movement disorders (MDs) with life-threatening and drug-resistant paroxysmal exacerbations, neurodevelopmental disorders, and epilepsy. Recently, the phenotypic spectrum has broadened to include milder phenotypes with late-onset dystonia, minor cognitive impairment, and other neurological signs, including parkinsonism and myoclonus. <i>GNAO1</i> haploinsufficiency has been evoked as a putative mechanism underlying milder clinical presentations.<span><sup>1, 2</sup></span> To date, however, the functional consequences of this class of variants have not yet been evaluated.</p><p>We report on an 8-year-old boy with subtle neurological signs, including generalized tonic–clonic seizures during fever, mild language impairment, dystonic postures of lower limbs during walking, and occasional tongue dyskinetic movements (see Data S1 for more details and Video 1). A next generation sequencing–based epilepsy panel revealed a de novo NM_020988.3:c.163_164del variant in <i>GNAO1</i>. No additional candidate variants were identified. Reverse transcription polymerase chain reaction showed an approximately 50% decrease in the expression of the endogenous <i>GNAO1</i> gene in cells from the affected child compared with cells from the unaffected father (Figs. 1A and S2), suggesting nonsense-mediated mRNA decay (NMD). If translated, the c.163_164delAT allele was predicted to generate a truncated protein (p.Ile55Hisfs*3). As expected, Western blotting performed in transiently transfected HEK293T cells revealed the lack of the truncated form of Gαo (Fig. 1B), which was not restored by MG132 or bafilomycin treatments, inhibitors of the ubiquitin/proteasome and autophagy pathways, respectively. These findings demonstrate that the c.163_164delAT transcript undergoes NMD, leading to <i>GNAO1</i> haploinsufficiency.</p><p>Genotype/phenotype correlations in <i>GNAO1</i> encephalopathy are still far from being elucidated. Recent studies suggest that pathogenic variants have a loss-of-function effect on Gαo-mediated signaling,<span><sup>3-7</sup></span> but the consequences on G-beta-gamma subunit (Gβγ) signaling that regulates cyclic adenosine monophosphate production remain unclear. Emerging data show that haploinsufficiency is associated with milder clinical features and later onset than missense changes underlying developmental and epileptic encephalopathy type 17 (Mendelian inheritance in man [MIM]#615473) or neurodevelopmental disorder with involuntary movements (MIM#617493). This finding has important implications. First, given the different phenotypic output, variants associated with the canonical form of <i>GNAO1</i> encephalopathy cannot have a simple loss-of-function effect; rather, they behave as dominant-negative alleles or alter Gα/Gβγ association, as recently shown for a subset of changes.<span><sup>3-7</sup></span> Second, the phenotype associated with <i>GNAO1</i> haploinsufficiency is likely attributed to increased levels of free Gβγ in the brain, which, in turn, could lead to increased receptor-independent Gβγ signaling in neurons. Finally, the association of <i>GNAO1</i> haploinsufficiency with a subtle but distinctive phenotype may help to design a proper gene therapy strategy. Allele-specific silencing by antisense oligonucleotides or short-interfering RNAs is unlikely to be a reasonable approach because 50% of the gene dosage is not neutral and single nucleotide substitutions hardly confer a complete discrimination for allele-specific targeting. In contrast, AAV-mediated gene supplementation coupled with silencing of the mutant allele is expected to effectively alleviate the disease phenotype.</p><p>Our findings also indicate that <i>GNAO1</i> variants may be more frequent than previously estimated and encourage testing for this gene in patients with mild neurological signs featuring epilepsy and/or MDs without a definite diagnosis. The progression into more severe phenotypes, and possible neurological deterioration induced by triggering events, typical for this condition, deserve a careful and prolonged clinical follow-up.</p><p>(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.</p><p>S.G.: 1A, 1B, 2B, 2C, 3A, 3B</p><p>M.N.: 1A, 1B, 3A, 3B</p><p>M.R.: 2B, 2C</p><p>E.F.: 3B</p><p>E.M.: 3B</p><p>L.P.: 3B</p><p>E.P.: 2B, 2C</p><p>F.P.: 3B</p><p>R.G.: 3B</p><p>V.L.: 1A, 1B, 3A, 3B</p><p>S.M.: 1A, 1B, 2B, 2C, 3A, 3B</p><p>None.</p>\",\"PeriodicalId\":213,\"journal\":{\"name\":\"Movement Disorders\",\"volume\":\"38 12\",\"pages\":\"2313-2314\"},\"PeriodicalIF\":7.4000,\"publicationDate\":\"2023-08-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://movementdisorders.onlinelibrary.wiley.com/doi/epdf/10.1002/mds.29585\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Movement Disorders\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/mds.29585\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Movement Disorders","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/mds.29585","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
GNAO1 Haploinsufficiency: The Milder End of the GNAO1 Phenotypic Spectrum
GNAO1 variants are typically associated with severe, early-onset movement disorders (MDs) with life-threatening and drug-resistant paroxysmal exacerbations, neurodevelopmental disorders, and epilepsy. Recently, the phenotypic spectrum has broadened to include milder phenotypes with late-onset dystonia, minor cognitive impairment, and other neurological signs, including parkinsonism and myoclonus. GNAO1 haploinsufficiency has been evoked as a putative mechanism underlying milder clinical presentations.1, 2 To date, however, the functional consequences of this class of variants have not yet been evaluated.
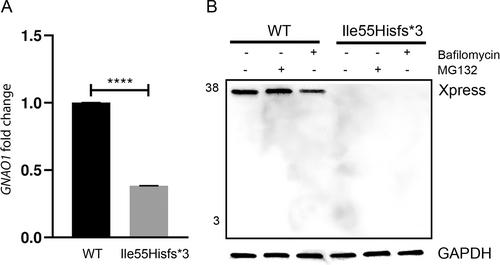
We report on an 8-year-old boy with subtle neurological signs, including generalized tonic–clonic seizures during fever, mild language impairment, dystonic postures of lower limbs during walking, and occasional tongue dyskinetic movements (see Data S1 for more details and Video 1). A next generation sequencing–based epilepsy panel revealed a de novo NM_020988.3:c.163_164del variant in GNAO1. No additional candidate variants were identified. Reverse transcription polymerase chain reaction showed an approximately 50% decrease in the expression of the endogenous GNAO1 gene in cells from the affected child compared with cells from the unaffected father (Figs. 1A and S2), suggesting nonsense-mediated mRNA decay (NMD). If translated, the c.163_164delAT allele was predicted to generate a truncated protein (p.Ile55Hisfs*3). As expected, Western blotting performed in transiently transfected HEK293T cells revealed the lack of the truncated form of Gαo (Fig. 1B), which was not restored by MG132 or bafilomycin treatments, inhibitors of the ubiquitin/proteasome and autophagy pathways, respectively. These findings demonstrate that the c.163_164delAT transcript undergoes NMD, leading to GNAO1 haploinsufficiency.
Genotype/phenotype correlations in GNAO1 encephalopathy are still far from being elucidated. Recent studies suggest that pathogenic variants have a loss-of-function effect on Gαo-mediated signaling,3-7 but the consequences on G-beta-gamma subunit (Gβγ) signaling that regulates cyclic adenosine monophosphate production remain unclear. Emerging data show that haploinsufficiency is associated with milder clinical features and later onset than missense changes underlying developmental and epileptic encephalopathy type 17 (Mendelian inheritance in man [MIM]#615473) or neurodevelopmental disorder with involuntary movements (MIM#617493). This finding has important implications. First, given the different phenotypic output, variants associated with the canonical form of GNAO1 encephalopathy cannot have a simple loss-of-function effect; rather, they behave as dominant-negative alleles or alter Gα/Gβγ association, as recently shown for a subset of changes.3-7 Second, the phenotype associated with GNAO1 haploinsufficiency is likely attributed to increased levels of free Gβγ in the brain, which, in turn, could lead to increased receptor-independent Gβγ signaling in neurons. Finally, the association of GNAO1 haploinsufficiency with a subtle but distinctive phenotype may help to design a proper gene therapy strategy. Allele-specific silencing by antisense oligonucleotides or short-interfering RNAs is unlikely to be a reasonable approach because 50% of the gene dosage is not neutral and single nucleotide substitutions hardly confer a complete discrimination for allele-specific targeting. In contrast, AAV-mediated gene supplementation coupled with silencing of the mutant allele is expected to effectively alleviate the disease phenotype.
Our findings also indicate that GNAO1 variants may be more frequent than previously estimated and encourage testing for this gene in patients with mild neurological signs featuring epilepsy and/or MDs without a definite diagnosis. The progression into more severe phenotypes, and possible neurological deterioration induced by triggering events, typical for this condition, deserve a careful and prolonged clinical follow-up.
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
期刊介绍:
Movement Disorders publishes a variety of content types including Reviews, Viewpoints, Full Length Articles, Historical Reports, Brief Reports, and Letters. The journal considers original manuscripts on topics related to the diagnosis, therapeutics, pharmacology, biochemistry, physiology, etiology, genetics, and epidemiology of movement disorders. Appropriate topics include Parkinsonism, Chorea, Tremors, Dystonia, Myoclonus, Tics, Tardive Dyskinesia, Spasticity, and Ataxia.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


