Karlijn Bouman, Jan T Groothuis, Jonne Doorduin, Nens van Alfen, Floris E A Udink Ten Cate, Frederik M A van den Heuvel, Robin Nijveldt, Erik-Jan Kamsteeg, Anne T M Dittrich, Jos M T Draaisma, Mirian C H Janssen, Baziel G M van Engelen, Corrie E Erasmus, Nicol C Voermans
{"title":"LAMA2相关的终生肌肉营养不良:一项横断面研究。","authors":"Karlijn Bouman, Jan T Groothuis, Jonne Doorduin, Nens van Alfen, Floris E A Udink Ten Cate, Frederik M A van den Heuvel, Robin Nijveldt, Erik-Jan Kamsteeg, Anne T M Dittrich, Jos M T Draaisma, Mirian C H Janssen, Baziel G M van Engelen, Corrie E Erasmus, Nicol C Voermans","doi":"10.1212/NXG.0000000000200089","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong><i>LAMA2</i>-related muscular dystrophy (<i>LAMA2</i>-MD) is a rare neuromuscular disease characterized by proximal and axial muscle weakness, rigidity of the spine, scoliosis, and respiratory impairment. No curative treatment options exist, yet promising preclinical studies are ongoing. Currently, there is a paucity on natural history data, and appropriate clinical and functional outcome measures are needed. We aim for deep clinical phenotyping, establishment of a well-characterized baseline cohort for prospective follow-up and recruitment for future clinical trials, improvement of clinical care, and selection of outcome measures for reaching trial readiness.</p><p><strong>Methods: </strong>We performed a cross-sectional, single-center, observational study. This study included neurologic examination and functional measurements among others the Motor Function Measure 20/32 (MFM-20/32) as primary outcome measure, accelerometry, questionnaires, muscle ultrasound, respiratory function tests, electrocardiography and echocardiography, and dual-energy X-ray absorptiometry.</p><p><strong>Results: </strong>Twenty-seven patients with genetically confirmed <i>LAMA2</i>-MD were included (21 ± 13 years; M = 9; ambulant = 7). Axial and proximal muscle weakness was most pronounced. The mean MFM-20/32 score was 42.0% ± 29.4%, with domain 1 (standing and transfers) being severely affected and domain 3 (distal muscle function) relatively spared. Physical activity as measured through accelerometry showed very strong correlations to MFM-20/32 (Pearson correlation, -0.928, <i>p</i> < 0.01). Muscle ultrasound showed symmetrically increased echogenicity, with the sternocleidomastoid muscle most affected. Respiratory function was impaired in 85% of patients without prominent diaphragm dysfunction and was independent of age. Ten patients (37%) needed (non)invasive ventilatory support. Cardiac assessment revealed QRS fragmentation in 62%, abnormal left ventricular global longitudinal strain in 25%, and decreased left ventricular ejection fraction in 14% of patients. Decreased bone quality leading to fragility fractures was seen in most of the patients.</p><p><strong>Discussion: </strong><i>LAMA2</i>-MD has a widely variable phenotype. Based on the results of this cross-sectional study and current standards of care for congenital muscular dystrophies, we advise routine cardiorespiratory follow-up and optimization of bone quality. We propose MFM-20/32, accelerometry, and muscle ultrasound for assessing disease severity and progression. For definitive clinical recommendations and outcome measures, natural history data are needed.</p><p><strong>Clinical trials registration: </strong>This study was registered at clinicaltrials.gov (NCT04478981, 21 July 2020). The first patient was enrolled in September 2020.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"9 5","pages":"e200089"},"PeriodicalIF":3.0000,"publicationDate":"2023-07-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/01/98/NXG-2023-000029.PMC10356133.pdf","citationCount":"3","resultStr":"{\"title\":\"<i>LAMA2</i>-Related Muscular Dystrophy Across the Life Span: A Cross-sectional Study.\",\"authors\":\"Karlijn Bouman, Jan T Groothuis, Jonne Doorduin, Nens van Alfen, Floris E A Udink Ten Cate, Frederik M A van den Heuvel, Robin Nijveldt, Erik-Jan Kamsteeg, Anne T M Dittrich, Jos M T Draaisma, Mirian C H Janssen, Baziel G M van Engelen, Corrie E Erasmus, Nicol C Voermans\",\"doi\":\"10.1212/NXG.0000000000200089\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background and objectives: </strong><i>LAMA2</i>-related muscular dystrophy (<i>LAMA2</i>-MD) is a rare neuromuscular disease characterized by proximal and axial muscle weakness, rigidity of the spine, scoliosis, and respiratory impairment. No curative treatment options exist, yet promising preclinical studies are ongoing. Currently, there is a paucity on natural history data, and appropriate clinical and functional outcome measures are needed. We aim for deep clinical phenotyping, establishment of a well-characterized baseline cohort for prospective follow-up and recruitment for future clinical trials, improvement of clinical care, and selection of outcome measures for reaching trial readiness.</p><p><strong>Methods: </strong>We performed a cross-sectional, single-center, observational study. This study included neurologic examination and functional measurements among others the Motor Function Measure 20/32 (MFM-20/32) as primary outcome measure, accelerometry, questionnaires, muscle ultrasound, respiratory function tests, electrocardiography and echocardiography, and dual-energy X-ray absorptiometry.</p><p><strong>Results: </strong>Twenty-seven patients with genetically confirmed <i>LAMA2</i>-MD were included (21 ± 13 years; M = 9; ambulant = 7). Axial and proximal muscle weakness was most pronounced. The mean MFM-20/32 score was 42.0% ± 29.4%, with domain 1 (standing and transfers) being severely affected and domain 3 (distal muscle function) relatively spared. Physical activity as measured through accelerometry showed very strong correlations to MFM-20/32 (Pearson correlation, -0.928, <i>p</i> < 0.01). Muscle ultrasound showed symmetrically increased echogenicity, with the sternocleidomastoid muscle most affected. Respiratory function was impaired in 85% of patients without prominent diaphragm dysfunction and was independent of age. Ten patients (37%) needed (non)invasive ventilatory support. Cardiac assessment revealed QRS fragmentation in 62%, abnormal left ventricular global longitudinal strain in 25%, and decreased left ventricular ejection fraction in 14% of patients. Decreased bone quality leading to fragility fractures was seen in most of the patients.</p><p><strong>Discussion: </strong><i>LAMA2</i>-MD has a widely variable phenotype. Based on the results of this cross-sectional study and current standards of care for congenital muscular dystrophies, we advise routine cardiorespiratory follow-up and optimization of bone quality. We propose MFM-20/32, accelerometry, and muscle ultrasound for assessing disease severity and progression. For definitive clinical recommendations and outcome measures, natural history data are needed.</p><p><strong>Clinical trials registration: </strong>This study was registered at clinicaltrials.gov (NCT04478981, 21 July 2020). The first patient was enrolled in September 2020.</p>\",\"PeriodicalId\":48613,\"journal\":{\"name\":\"Neurology-Genetics\",\"volume\":\"9 5\",\"pages\":\"e200089\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2023-07-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/01/98/NXG-2023-000029.PMC10356133.pdf\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurology-Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1212/NXG.0000000000200089\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/10/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200089","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/10/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
LAMA2-Related Muscular Dystrophy Across the Life Span: A Cross-sectional Study.
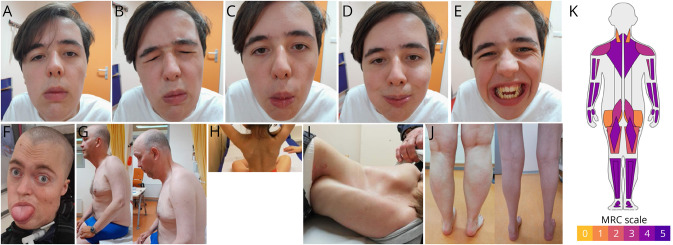
Background and objectives: LAMA2-related muscular dystrophy (LAMA2-MD) is a rare neuromuscular disease characterized by proximal and axial muscle weakness, rigidity of the spine, scoliosis, and respiratory impairment. No curative treatment options exist, yet promising preclinical studies are ongoing. Currently, there is a paucity on natural history data, and appropriate clinical and functional outcome measures are needed. We aim for deep clinical phenotyping, establishment of a well-characterized baseline cohort for prospective follow-up and recruitment for future clinical trials, improvement of clinical care, and selection of outcome measures for reaching trial readiness.
Methods: We performed a cross-sectional, single-center, observational study. This study included neurologic examination and functional measurements among others the Motor Function Measure 20/32 (MFM-20/32) as primary outcome measure, accelerometry, questionnaires, muscle ultrasound, respiratory function tests, electrocardiography and echocardiography, and dual-energy X-ray absorptiometry.
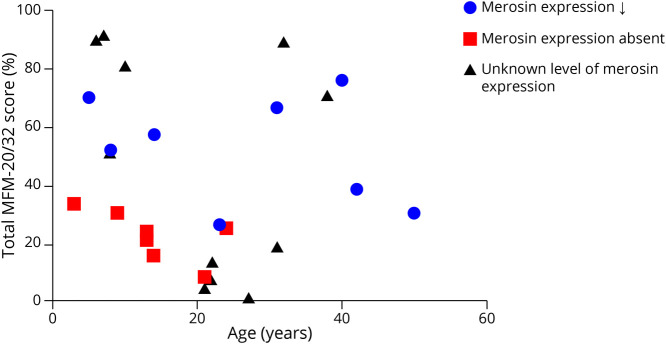
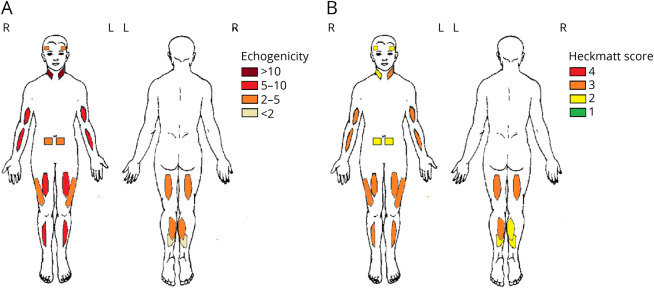
Results: Twenty-seven patients with genetically confirmed LAMA2-MD were included (21 ± 13 years; M = 9; ambulant = 7). Axial and proximal muscle weakness was most pronounced. The mean MFM-20/32 score was 42.0% ± 29.4%, with domain 1 (standing and transfers) being severely affected and domain 3 (distal muscle function) relatively spared. Physical activity as measured through accelerometry showed very strong correlations to MFM-20/32 (Pearson correlation, -0.928, p < 0.01). Muscle ultrasound showed symmetrically increased echogenicity, with the sternocleidomastoid muscle most affected. Respiratory function was impaired in 85% of patients without prominent diaphragm dysfunction and was independent of age. Ten patients (37%) needed (non)invasive ventilatory support. Cardiac assessment revealed QRS fragmentation in 62%, abnormal left ventricular global longitudinal strain in 25%, and decreased left ventricular ejection fraction in 14% of patients. Decreased bone quality leading to fragility fractures was seen in most of the patients.
Discussion: LAMA2-MD has a widely variable phenotype. Based on the results of this cross-sectional study and current standards of care for congenital muscular dystrophies, we advise routine cardiorespiratory follow-up and optimization of bone quality. We propose MFM-20/32, accelerometry, and muscle ultrasound for assessing disease severity and progression. For definitive clinical recommendations and outcome measures, natural history data are needed.
Clinical trials registration: This study was registered at clinicaltrials.gov (NCT04478981, 21 July 2020). The first patient was enrolled in September 2020.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


