Mark P. Simmons, Pablo A. Goloboff, Ben C. Stöver, Mark S. Springer, John Gatesy
{"title":"利用系统发育学联合分析的整体成功解析来量化基因树与多面体之间的一致性。","authors":"Mark P. Simmons, Pablo A. Goloboff, Ben C. Stöver, Mark S. Springer, John Gatesy","doi":"10.1111/cla.12540","DOIUrl":null,"url":null,"abstract":"<p>Gene-tree-inference error can cause species-tree-inference artefacts in summary phylogenomic coalescent analyses. Here we integrate two ways of accommodating these inference errors: collapsing arbitrarily or dubiously resolved gene-tree branches, and subsampling gene trees based on their pairwise congruence. We tested the effect of collapsing gene-tree branches with 0% approximate-likelihood-ratio-test (SH-like aLRT) support in likelihood analyses and strict consensus trees for parsimony, and then subsampled those partially resolved trees based on congruence measures that do not penalize polytomies. For this purpose we developed a new TNT script for congruence sorting (<span>congsort</span>), and used it to calculate topological incongruence for eight phylogenomic datasets using three distance measures: standard Robinson–Foulds (RF) distances; overall success of resolution (OSR), which is based on counting both matching and contradicting clades; and RF contradictions, which only counts contradictory clades. As expected, we found that gene-tree incongruence was often concentrated in clades that are arbitrarily or dubiously resolved and that there was greater congruence between the partially collapsed gene trees and the coalescent and concatenation topologies inferred from those genes. Coalescent branch lengths typically increased as the most incongruent gene trees were excluded, although branch supports typically did not. We investigated two successful and complementary approaches to prioritizing genes for investigation of alignment or homology errors. Coalescent-tree clades that contradicted concatenation-tree clades were generally less robust to gene-tree subsampling than congruent clades. Our preferred approach to collapsing likelihood gene-tree clades (0% SH-like aLRT support) and subsampling those trees (OSR) generally outperformed competing approaches for a large fungal dataset with respect to branch lengths, support and congruence. We recommend widespread application of this approach (and strict consensus trees for parsimony-based analyses) for improving quantification of gene-tree congruence/conflict, estimating coalescent branch lengths, testing robustness of coalescent analyses to gene-tree-estimation error, and improving topological robustness of summary coalescent analyses. This approach is quick and easy to implement, even for huge datasets.</p>","PeriodicalId":50688,"journal":{"name":"Cladistics","volume":"39 5","pages":"418-436"},"PeriodicalIF":6.2000,"publicationDate":"2023-04-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cla.12540","citationCount":"1","resultStr":"{\"title\":\"Quantification of congruence among gene trees with polytomies using overall success of resolution for phylogenomic coalescent analyses\",\"authors\":\"Mark P. Simmons, Pablo A. Goloboff, Ben C. Stöver, Mark S. Springer, John Gatesy\",\"doi\":\"10.1111/cla.12540\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Gene-tree-inference error can cause species-tree-inference artefacts in summary phylogenomic coalescent analyses. Here we integrate two ways of accommodating these inference errors: collapsing arbitrarily or dubiously resolved gene-tree branches, and subsampling gene trees based on their pairwise congruence. We tested the effect of collapsing gene-tree branches with 0% approximate-likelihood-ratio-test (SH-like aLRT) support in likelihood analyses and strict consensus trees for parsimony, and then subsampled those partially resolved trees based on congruence measures that do not penalize polytomies. For this purpose we developed a new TNT script for congruence sorting (<span>congsort</span>), and used it to calculate topological incongruence for eight phylogenomic datasets using three distance measures: standard Robinson–Foulds (RF) distances; overall success of resolution (OSR), which is based on counting both matching and contradicting clades; and RF contradictions, which only counts contradictory clades. As expected, we found that gene-tree incongruence was often concentrated in clades that are arbitrarily or dubiously resolved and that there was greater congruence between the partially collapsed gene trees and the coalescent and concatenation topologies inferred from those genes. Coalescent branch lengths typically increased as the most incongruent gene trees were excluded, although branch supports typically did not. We investigated two successful and complementary approaches to prioritizing genes for investigation of alignment or homology errors. Coalescent-tree clades that contradicted concatenation-tree clades were generally less robust to gene-tree subsampling than congruent clades. Our preferred approach to collapsing likelihood gene-tree clades (0% SH-like aLRT support) and subsampling those trees (OSR) generally outperformed competing approaches for a large fungal dataset with respect to branch lengths, support and congruence. We recommend widespread application of this approach (and strict consensus trees for parsimony-based analyses) for improving quantification of gene-tree congruence/conflict, estimating coalescent branch lengths, testing robustness of coalescent analyses to gene-tree-estimation error, and improving topological robustness of summary coalescent analyses. This approach is quick and easy to implement, even for huge datasets.</p>\",\"PeriodicalId\":50688,\"journal\":{\"name\":\"Cladistics\",\"volume\":\"39 5\",\"pages\":\"418-436\"},\"PeriodicalIF\":6.2000,\"publicationDate\":\"2023-04-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cla.12540\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cladistics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/cla.12540\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"EVOLUTIONARY BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cladistics","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cla.12540","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"EVOLUTIONARY BIOLOGY","Score":null,"Total":0}
Quantification of congruence among gene trees with polytomies using overall success of resolution for phylogenomic coalescent analyses
Gene-tree-inference error can cause species-tree-inference artefacts in summary phylogenomic coalescent analyses. Here we integrate two ways of accommodating these inference errors: collapsing arbitrarily or dubiously resolved gene-tree branches, and subsampling gene trees based on their pairwise congruence. We tested the effect of collapsing gene-tree branches with 0% approximate-likelihood-ratio-test (SH-like aLRT) support in likelihood analyses and strict consensus trees for parsimony, and then subsampled those partially resolved trees based on congruence measures that do not penalize polytomies. For this purpose we developed a new TNT script for congruence sorting (congsort), and used it to calculate topological incongruence for eight phylogenomic datasets using three distance measures: standard Robinson–Foulds (RF) distances; overall success of resolution (OSR), which is based on counting both matching and contradicting clades; and RF contradictions, which only counts contradictory clades. As expected, we found that gene-tree incongruence was often concentrated in clades that are arbitrarily or dubiously resolved and that there was greater congruence between the partially collapsed gene trees and the coalescent and concatenation topologies inferred from those genes. Coalescent branch lengths typically increased as the most incongruent gene trees were excluded, although branch supports typically did not. We investigated two successful and complementary approaches to prioritizing genes for investigation of alignment or homology errors. Coalescent-tree clades that contradicted concatenation-tree clades were generally less robust to gene-tree subsampling than congruent clades. Our preferred approach to collapsing likelihood gene-tree clades (0% SH-like aLRT support) and subsampling those trees (OSR) generally outperformed competing approaches for a large fungal dataset with respect to branch lengths, support and congruence. We recommend widespread application of this approach (and strict consensus trees for parsimony-based analyses) for improving quantification of gene-tree congruence/conflict, estimating coalescent branch lengths, testing robustness of coalescent analyses to gene-tree-estimation error, and improving topological robustness of summary coalescent analyses. This approach is quick and easy to implement, even for huge datasets.
期刊介绍:
Cladistics publishes high quality research papers on systematics, encouraging debate on all aspects of the field, from philosophy, theory and methodology to empirical studies and applications in biogeography, coevolution, conservation biology, ontogeny, genomics and paleontology.
Cladistics is read by scientists working in the research fields of evolution, systematics and integrative biology and enjoys a consistently high position in the ISI® rankings for evolutionary biology.
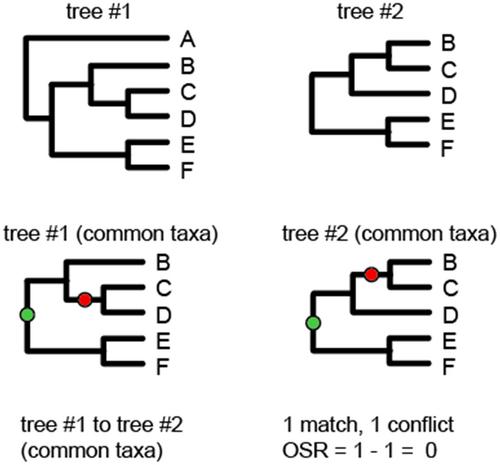

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


