{"title":"利用参考文献指导的组装和硅库改进了蝠鲼和 Culter alburnus 的基因组草案。","authors":"Kai Liu, Nan Xie, Yuxi Wang, Xinyi Liu","doi":"10.1007/s10126-023-10248-x","DOIUrl":null,"url":null,"abstract":"<div><p>Long-read sequencing technologies can generate highly contiguous genome assemblies compared to short-read methods. However, their higher cost often poses a significant barrier. To address this, we explore the utilization of mapping-based genome assembly and reference-guided assembly as cost-effective alternative approaches. We assess the efficacy of these approaches in improving the contiguity of <i>Clarias batrachus</i> and <i>Culter alburnus</i> draft genomes. Our findings demonstrate that employing an iterative mapping strategy leads to a reduction in assembly errors. Specifically, after three iterations, the Mismatches per 100 kbp value for the <i>C. batrachus</i> genome decreased from 2447.20 to 2432.67, reaching a minimum of 2422.67 after two iterations. Additionally, the N50 value for the <i>C. batrachus</i> genome increased from 362,143 to 1,315,126 bp, with a maximum of 1,315,403 bp after two iterations. Furthermore, we achieved Mismatches per 100 kbp values of 3.70 for the reference-guided assembly of <i>C. batrachus</i> and 0.34 for <i>C. alburnus</i>. Correspondingly, the N50 value for the <i>C. batrachus</i> and <i>C. alburnus</i> genomes increased from 362,143 bp and 3,686,385 bp to 2,026,888 bp and 43,735,735 bp, respectively. Finally, we successfully utilized the improved <i>C. batrachus</i> and <i>C. alburnus</i> genomes to compare genome studies using the combined approach of Ragout and Ragtag. Through a comprehensive comparative analysis of mapping-based and reference-guided genome assembly methods, we shed light on the specific contributions of reference-guided assembly in reducing assembly errors and improving assembly continuity and integrity. These advancements establish reference-guided assembly and the utilization of in silico libraries as a promising and suitable approach for comparative genomics studies.</p></div>","PeriodicalId":690,"journal":{"name":"Marine Biotechnology","volume":"25 6","pages":"907 - 917"},"PeriodicalIF":2.6000,"publicationDate":"2023-09-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"The Utilization of Reference-Guided Assembly and In Silico Libraries Improves the Draft Genome of Clarias batrachus and Culter alburnus\",\"authors\":\"Kai Liu, Nan Xie, Yuxi Wang, Xinyi Liu\",\"doi\":\"10.1007/s10126-023-10248-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Long-read sequencing technologies can generate highly contiguous genome assemblies compared to short-read methods. However, their higher cost often poses a significant barrier. To address this, we explore the utilization of mapping-based genome assembly and reference-guided assembly as cost-effective alternative approaches. We assess the efficacy of these approaches in improving the contiguity of <i>Clarias batrachus</i> and <i>Culter alburnus</i> draft genomes. Our findings demonstrate that employing an iterative mapping strategy leads to a reduction in assembly errors. Specifically, after three iterations, the Mismatches per 100 kbp value for the <i>C. batrachus</i> genome decreased from 2447.20 to 2432.67, reaching a minimum of 2422.67 after two iterations. Additionally, the N50 value for the <i>C. batrachus</i> genome increased from 362,143 to 1,315,126 bp, with a maximum of 1,315,403 bp after two iterations. Furthermore, we achieved Mismatches per 100 kbp values of 3.70 for the reference-guided assembly of <i>C. batrachus</i> and 0.34 for <i>C. alburnus</i>. Correspondingly, the N50 value for the <i>C. batrachus</i> and <i>C. alburnus</i> genomes increased from 362,143 bp and 3,686,385 bp to 2,026,888 bp and 43,735,735 bp, respectively. Finally, we successfully utilized the improved <i>C. batrachus</i> and <i>C. alburnus</i> genomes to compare genome studies using the combined approach of Ragout and Ragtag. Through a comprehensive comparative analysis of mapping-based and reference-guided genome assembly methods, we shed light on the specific contributions of reference-guided assembly in reducing assembly errors and improving assembly continuity and integrity. These advancements establish reference-guided assembly and the utilization of in silico libraries as a promising and suitable approach for comparative genomics studies.</p></div>\",\"PeriodicalId\":690,\"journal\":{\"name\":\"Marine Biotechnology\",\"volume\":\"25 6\",\"pages\":\"907 - 917\"},\"PeriodicalIF\":2.6000,\"publicationDate\":\"2023-09-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Marine Biotechnology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10126-023-10248-x\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOTECHNOLOGY & APPLIED MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Marine Biotechnology","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10126-023-10248-x","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
The Utilization of Reference-Guided Assembly and In Silico Libraries Improves the Draft Genome of Clarias batrachus and Culter alburnus
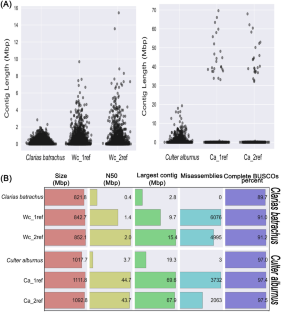
Long-read sequencing technologies can generate highly contiguous genome assemblies compared to short-read methods. However, their higher cost often poses a significant barrier. To address this, we explore the utilization of mapping-based genome assembly and reference-guided assembly as cost-effective alternative approaches. We assess the efficacy of these approaches in improving the contiguity of Clarias batrachus and Culter alburnus draft genomes. Our findings demonstrate that employing an iterative mapping strategy leads to a reduction in assembly errors. Specifically, after three iterations, the Mismatches per 100 kbp value for the C. batrachus genome decreased from 2447.20 to 2432.67, reaching a minimum of 2422.67 after two iterations. Additionally, the N50 value for the C. batrachus genome increased from 362,143 to 1,315,126 bp, with a maximum of 1,315,403 bp after two iterations. Furthermore, we achieved Mismatches per 100 kbp values of 3.70 for the reference-guided assembly of C. batrachus and 0.34 for C. alburnus. Correspondingly, the N50 value for the C. batrachus and C. alburnus genomes increased from 362,143 bp and 3,686,385 bp to 2,026,888 bp and 43,735,735 bp, respectively. Finally, we successfully utilized the improved C. batrachus and C. alburnus genomes to compare genome studies using the combined approach of Ragout and Ragtag. Through a comprehensive comparative analysis of mapping-based and reference-guided genome assembly methods, we shed light on the specific contributions of reference-guided assembly in reducing assembly errors and improving assembly continuity and integrity. These advancements establish reference-guided assembly and the utilization of in silico libraries as a promising and suitable approach for comparative genomics studies.
期刊介绍:
Marine Biotechnology welcomes high-quality research papers presenting novel data on the biotechnology of aquatic organisms. The journal publishes high quality papers in the areas of molecular biology, genomics, proteomics, cell biology, and biochemistry, and particularly encourages submissions of papers related to genome biology such as linkage mapping, large-scale gene discoveries, QTL analysis, physical mapping, and comparative and functional genome analysis. Papers on technological development and marine natural products should demonstrate innovation and novel applications.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


