Marion Almes, Antoine Gardin, Anne Davit-Spraul, Jérôme Bouligand, Dalila Habes, Emmanuel Jacquemin
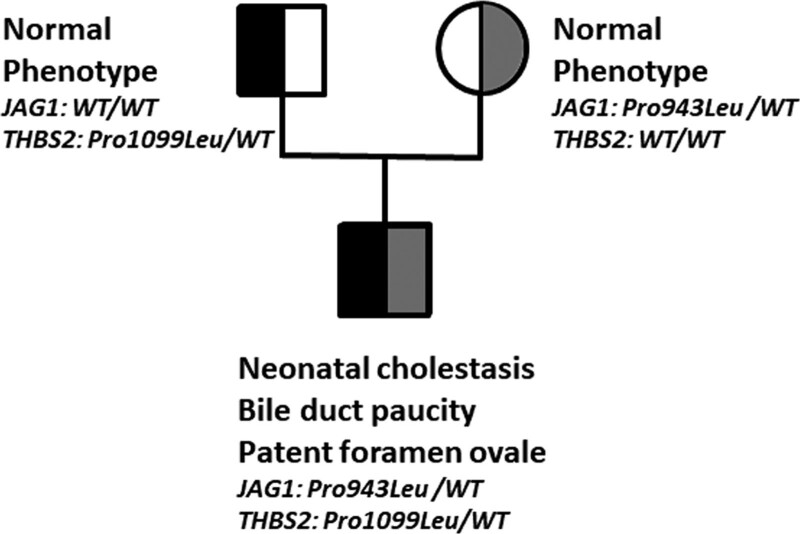
{"title":"不完全阿拉吉尔综合征患儿的JAG1和THBS2突变","authors":"Marion Almes, Antoine Gardin, Anne Davit-Spraul, Jérôme Bouligand, Dalila Habes, Emmanuel Jacquemin","doi":"10.1097/PG9.0000000000000338","DOIUrl":null,"url":null,"abstract":"TO THE EDITOR: Alagille syndrome (ALGS) is an autosomal-dominant multisystem disorder mainly caused by mutations in JAG1 (1,2). Phenotypic expressivity is variable and JAG1 mutations have been found in patients with incomplete ALGS presenting with <3 major features of the syndrome (3,4). Thus, it is also useful to look for a genetic cause in such ALGS-like patients. A genome-wide association study performed in ALGS patients with JAG1 mutations identified a significant locus upstream of the thrombospondin 2 gene (THBS2) (2). THBS2 regulates cell fate and angiogenesis and is expressed in bile ducts in mouse and human livers (2,5). THBS2 modifies JAG1–NOTCH2 interactions in vitro and a THBS2 SNP is associated with cardiovascular diseases (2,6,7). Therefore, THBS2 could be a candidate genetic modifier in ALGS patients, by disrupting JAG1–NOTCH2 signaling (2). So far, THBS2 mutations have not been reported in ALGS patients. We report on a boy who had neonatal cholestasis with elevated serum GGT activity (308 IU/L). At age of 2 months, cholangiography excluded biliary atresia, liver histology showed severe ductopenia, and cardiac ultrasonography a patent foramen ovale. Other target organs were not affected. Genetic analysis identified in the boy a maternal heterozygous JAG1 mutation (NM_000214; c.2828C>T; p.Pro943Leu; ACMG classification: class 3; gnomAD 0.00438%) and a paternal heterozygous THBS2 mutation (NM_003247; c.3296C>T; p.Pro1099Leu; class 2; gnomAD 0.009%) (Fig. 1). His parents were healthy but the mother who transmitted the JAG1 mutation refused any clinical investigation. This observation in a child with incomplete ALGS affecting only the liver and heart, together with data from the literature (2), further suggests that THBS2 could be a modifier gene in some ALGS patients with JAG1 mutations and could play a role in the variable expressivity of this syndrome. The combination of both mutants could explain the incomplete ALGS phenotype in the propositus, by disrupting the JAG1–NOTCH2 signaling. However, a functional evaluation of the interaction of a THBS2 mutant on the Notch signaling pathway as well as the search for THBS2 mutations in patients with ALGS are needed to conclude that THBS2 can be a modifier gene in ALGS.","PeriodicalId":17618,"journal":{"name":"JPGN Reports","volume":"4 3","pages":"e338"},"PeriodicalIF":0.0000,"publicationDate":"2023-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10435021/pdf/","citationCount":"0","resultStr":"{\"title\":\"<i>JAG1</i> and <i>THBS2</i> Mutations in a Child Presenting With Incomplete Alagille Syndrome.\",\"authors\":\"Marion Almes, Antoine Gardin, Anne Davit-Spraul, Jérôme Bouligand, Dalila Habes, Emmanuel Jacquemin\",\"doi\":\"10.1097/PG9.0000000000000338\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"TO THE EDITOR: Alagille syndrome (ALGS) is an autosomal-dominant multisystem disorder mainly caused by mutations in JAG1 (1,2). Phenotypic expressivity is variable and JAG1 mutations have been found in patients with incomplete ALGS presenting with <3 major features of the syndrome (3,4). Thus, it is also useful to look for a genetic cause in such ALGS-like patients. A genome-wide association study performed in ALGS patients with JAG1 mutations identified a significant locus upstream of the thrombospondin 2 gene (THBS2) (2). THBS2 regulates cell fate and angiogenesis and is expressed in bile ducts in mouse and human livers (2,5). THBS2 modifies JAG1–NOTCH2 interactions in vitro and a THBS2 SNP is associated with cardiovascular diseases (2,6,7). Therefore, THBS2 could be a candidate genetic modifier in ALGS patients, by disrupting JAG1–NOTCH2 signaling (2). So far, THBS2 mutations have not been reported in ALGS patients. We report on a boy who had neonatal cholestasis with elevated serum GGT activity (308 IU/L). At age of 2 months, cholangiography excluded biliary atresia, liver histology showed severe ductopenia, and cardiac ultrasonography a patent foramen ovale. Other target organs were not affected. Genetic analysis identified in the boy a maternal heterozygous JAG1 mutation (NM_000214; c.2828C>T; p.Pro943Leu; ACMG classification: class 3; gnomAD 0.00438%) and a paternal heterozygous THBS2 mutation (NM_003247; c.3296C>T; p.Pro1099Leu; class 2; gnomAD 0.009%) (Fig. 1). His parents were healthy but the mother who transmitted the JAG1 mutation refused any clinical investigation. This observation in a child with incomplete ALGS affecting only the liver and heart, together with data from the literature (2), further suggests that THBS2 could be a modifier gene in some ALGS patients with JAG1 mutations and could play a role in the variable expressivity of this syndrome. The combination of both mutants could explain the incomplete ALGS phenotype in the propositus, by disrupting the JAG1–NOTCH2 signaling. However, a functional evaluation of the interaction of a THBS2 mutant on the Notch signaling pathway as well as the search for THBS2 mutations in patients with ALGS are needed to conclude that THBS2 can be a modifier gene in ALGS.\",\"PeriodicalId\":17618,\"journal\":{\"name\":\"JPGN Reports\",\"volume\":\"4 3\",\"pages\":\"e338\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-08-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10435021/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JPGN Reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1097/PG9.0000000000000338\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JPGN Reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1097/PG9.0000000000000338","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
JAG1 and THBS2 Mutations in a Child Presenting With Incomplete Alagille Syndrome.
TO THE EDITOR: Alagille syndrome (ALGS) is an autosomal-dominant multisystem disorder mainly caused by mutations in JAG1 (1,2). Phenotypic expressivity is variable and JAG1 mutations have been found in patients with incomplete ALGS presenting with <3 major features of the syndrome (3,4). Thus, it is also useful to look for a genetic cause in such ALGS-like patients. A genome-wide association study performed in ALGS patients with JAG1 mutations identified a significant locus upstream of the thrombospondin 2 gene (THBS2) (2). THBS2 regulates cell fate and angiogenesis and is expressed in bile ducts in mouse and human livers (2,5). THBS2 modifies JAG1–NOTCH2 interactions in vitro and a THBS2 SNP is associated with cardiovascular diseases (2,6,7). Therefore, THBS2 could be a candidate genetic modifier in ALGS patients, by disrupting JAG1–NOTCH2 signaling (2). So far, THBS2 mutations have not been reported in ALGS patients. We report on a boy who had neonatal cholestasis with elevated serum GGT activity (308 IU/L). At age of 2 months, cholangiography excluded biliary atresia, liver histology showed severe ductopenia, and cardiac ultrasonography a patent foramen ovale. Other target organs were not affected. Genetic analysis identified in the boy a maternal heterozygous JAG1 mutation (NM_000214; c.2828C>T; p.Pro943Leu; ACMG classification: class 3; gnomAD 0.00438%) and a paternal heterozygous THBS2 mutation (NM_003247; c.3296C>T; p.Pro1099Leu; class 2; gnomAD 0.009%) (Fig. 1). His parents were healthy but the mother who transmitted the JAG1 mutation refused any clinical investigation. This observation in a child with incomplete ALGS affecting only the liver and heart, together with data from the literature (2), further suggests that THBS2 could be a modifier gene in some ALGS patients with JAG1 mutations and could play a role in the variable expressivity of this syndrome. The combination of both mutants could explain the incomplete ALGS phenotype in the propositus, by disrupting the JAG1–NOTCH2 signaling. However, a functional evaluation of the interaction of a THBS2 mutant on the Notch signaling pathway as well as the search for THBS2 mutations in patients with ALGS are needed to conclude that THBS2 can be a modifier gene in ALGS.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


