Vidya S. Vuruputoor, Daniel Monyak, Karl C. Fetter, Cynthia Webster, Akriti Bhattarai, Bikash Shrestha, Sumaira Zaman, Jeremy Bennett, Susan L. McEvoy, Madison Caballero, Jill L. Wegrzyn
{"title":"欢迎来到大叶子:改进非模式植物基因组注释的最佳实践","authors":"Vidya S. Vuruputoor, Daniel Monyak, Karl C. Fetter, Cynthia Webster, Akriti Bhattarai, Bikash Shrestha, Sumaira Zaman, Jeremy Bennett, Susan L. McEvoy, Madison Caballero, Jill L. Wegrzyn","doi":"10.1002/aps3.11533","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Premise</h3>\n \n <p>Robust standards to evaluate quality and completeness are lacking in eukaryotic structural genome annotation, as genome annotation software is developed using model organisms and typically lacks benchmarking to comprehensively evaluate the quality and accuracy of the final predictions. The annotation of plant genomes is particularly challenging due to their large sizes, abundant transposable elements, and variable ploidies. This study investigates the impact of genome quality, complexity, sequence read input, and method on protein-coding gene predictions.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>The impact of repeat masking, long-read and short-read inputs, and de novo and genome-guided protein evidence was examined in the context of the popular BRAKER and MAKER workflows for five plant genomes. The annotations were benchmarked for structural traits and sequence similarity.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>Benchmarks that reflect gene structures, reciprocal similarity search alignments, and mono-exonic/multi-exonic gene counts provide a more complete view of annotation accuracy. Transcripts derived from RNA-read alignments alone are not sufficient for genome annotation. Gene prediction workflows that combine evidence-based and ab initio approaches are recommended, and a combination of short and long reads can improve genome annotation. Adding protein evidence from de novo assemblies, genome-guided transcriptome assemblies, or full-length proteins from OrthoDB generates more putative false positives as implemented in the current workflows. Post-processing with functional and structural filters is highly recommended.</p>\n </section>\n \n <section>\n \n <h3> Discussion</h3>\n \n <p>While the annotation of non-model plant genomes remains complex, this study provides recommendations for inputs and methodological approaches. We discuss a set of best practices to generate an optimal plant genome annotation and present a more robust set of metrics to evaluate the resulting predictions.</p>\n </section>\n </div>","PeriodicalId":8022,"journal":{"name":"Applications in Plant Sciences","volume":"11 4","pages":""},"PeriodicalIF":2.7000,"publicationDate":"2023-08-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://bsapubs.onlinelibrary.wiley.com/doi/epdf/10.1002/aps3.11533","citationCount":"6","resultStr":"{\"title\":\"Welcome to the big leaves: Best practices for improving genome annotation in non-model plant genomes\",\"authors\":\"Vidya S. Vuruputoor, Daniel Monyak, Karl C. Fetter, Cynthia Webster, Akriti Bhattarai, Bikash Shrestha, Sumaira Zaman, Jeremy Bennett, Susan L. McEvoy, Madison Caballero, Jill L. Wegrzyn\",\"doi\":\"10.1002/aps3.11533\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Premise</h3>\\n \\n <p>Robust standards to evaluate quality and completeness are lacking in eukaryotic structural genome annotation, as genome annotation software is developed using model organisms and typically lacks benchmarking to comprehensively evaluate the quality and accuracy of the final predictions. The annotation of plant genomes is particularly challenging due to their large sizes, abundant transposable elements, and variable ploidies. This study investigates the impact of genome quality, complexity, sequence read input, and method on protein-coding gene predictions.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>The impact of repeat masking, long-read and short-read inputs, and de novo and genome-guided protein evidence was examined in the context of the popular BRAKER and MAKER workflows for five plant genomes. The annotations were benchmarked for structural traits and sequence similarity.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>Benchmarks that reflect gene structures, reciprocal similarity search alignments, and mono-exonic/multi-exonic gene counts provide a more complete view of annotation accuracy. Transcripts derived from RNA-read alignments alone are not sufficient for genome annotation. Gene prediction workflows that combine evidence-based and ab initio approaches are recommended, and a combination of short and long reads can improve genome annotation. Adding protein evidence from de novo assemblies, genome-guided transcriptome assemblies, or full-length proteins from OrthoDB generates more putative false positives as implemented in the current workflows. Post-processing with functional and structural filters is highly recommended.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Discussion</h3>\\n \\n <p>While the annotation of non-model plant genomes remains complex, this study provides recommendations for inputs and methodological approaches. We discuss a set of best practices to generate an optimal plant genome annotation and present a more robust set of metrics to evaluate the resulting predictions.</p>\\n </section>\\n </div>\",\"PeriodicalId\":8022,\"journal\":{\"name\":\"Applications in Plant Sciences\",\"volume\":\"11 4\",\"pages\":\"\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2023-08-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://bsapubs.onlinelibrary.wiley.com/doi/epdf/10.1002/aps3.11533\",\"citationCount\":\"6\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Applications in Plant Sciences\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/aps3.11533\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"PLANT SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Applications in Plant Sciences","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/aps3.11533","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"PLANT SCIENCES","Score":null,"Total":0}
Welcome to the big leaves: Best practices for improving genome annotation in non-model plant genomes
Premise
Robust standards to evaluate quality and completeness are lacking in eukaryotic structural genome annotation, as genome annotation software is developed using model organisms and typically lacks benchmarking to comprehensively evaluate the quality and accuracy of the final predictions. The annotation of plant genomes is particularly challenging due to their large sizes, abundant transposable elements, and variable ploidies. This study investigates the impact of genome quality, complexity, sequence read input, and method on protein-coding gene predictions.
Methods
The impact of repeat masking, long-read and short-read inputs, and de novo and genome-guided protein evidence was examined in the context of the popular BRAKER and MAKER workflows for five plant genomes. The annotations were benchmarked for structural traits and sequence similarity.
Results
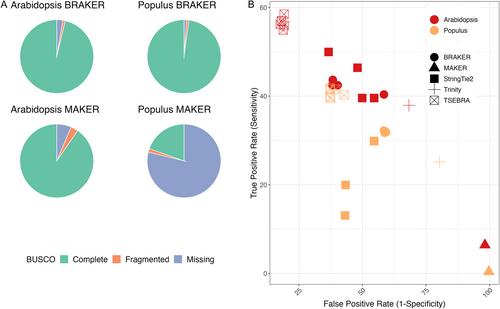
Benchmarks that reflect gene structures, reciprocal similarity search alignments, and mono-exonic/multi-exonic gene counts provide a more complete view of annotation accuracy. Transcripts derived from RNA-read alignments alone are not sufficient for genome annotation. Gene prediction workflows that combine evidence-based and ab initio approaches are recommended, and a combination of short and long reads can improve genome annotation. Adding protein evidence from de novo assemblies, genome-guided transcriptome assemblies, or full-length proteins from OrthoDB generates more putative false positives as implemented in the current workflows. Post-processing with functional and structural filters is highly recommended.
Discussion
While the annotation of non-model plant genomes remains complex, this study provides recommendations for inputs and methodological approaches. We discuss a set of best practices to generate an optimal plant genome annotation and present a more robust set of metrics to evaluate the resulting predictions.
期刊介绍:
Applications in Plant Sciences (APPS) is a monthly, peer-reviewed, open access journal promoting the rapid dissemination of newly developed, innovative tools and protocols in all areas of the plant sciences, including genetics, structure, function, development, evolution, systematics, and ecology. Given the rapid progress today in technology and its application in the plant sciences, the goal of APPS is to foster communication within the plant science community to advance scientific research. APPS is a publication of the Botanical Society of America, originating in 2009 as the American Journal of Botany''s online-only section, AJB Primer Notes & Protocols in the Plant Sciences.
APPS publishes the following types of articles: (1) Protocol Notes describe new methods and technological advancements; (2) Genomic Resources Articles characterize the development and demonstrate the usefulness of newly developed genomic resources, including transcriptomes; (3) Software Notes detail new software applications; (4) Application Articles illustrate the application of a new protocol, method, or software application within the context of a larger study; (5) Review Articles evaluate available techniques, methods, or protocols; (6) Primer Notes report novel genetic markers with evidence of wide applicability.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


