Edouard Palu, Julius Järvilehto, Jana Pennonen, Nadine Huber, Sanna-Kaisa Herukka, Annakaisa Haapasalo, Pirjo Isohanni, Henna Tyynismaa, Mari Auranen, Emil Ylikallio
{"title":"轻度至重度神经病变中罕见的PMP22变异与血浆GDF15或神经丝光无关。","authors":"Edouard Palu, Julius Järvilehto, Jana Pennonen, Nadine Huber, Sanna-Kaisa Herukka, Annakaisa Haapasalo, Pirjo Isohanni, Henna Tyynismaa, Mari Auranen, Emil Ylikallio","doi":"10.1007/s10048-023-00729-5","DOIUrl":null,"url":null,"abstract":"<p><p>Charcot-Marie-Tooth disease (CMT) is a heterogeneous set of hereditary neuropathies whose genetic causes are not fully understood. Here, we characterize three previously unknown variants in PMP22 and assess their effect on the recently described potential CMT biomarkers' growth differentiation factor 15 (GDF15) and neurofilament light (NFL): first, a heterozygous PMP22 c.178G > A (p.Glu60Lys) in one mother-son pair with adult-onset mild axonal neuropathy. The variant led to abnormal splicing, confirmed in fibroblasts by reverse transcription PCR. Second, a de novo PMP22 c.35A > C (p.His12Pro), and third, a heterozygous 3.2 kb deletion predicting loss of exon 4. The latter two had severe CMT and ultrasonography showing strong nerve enlargement similar to a previous case of exon 4 loss due to a larger deletion. We further studied patients with PMP22 duplication (CMT1A) finding slightly elevated plasma NFL, as measured by the single molecule array immunoassay (SIMOA). In addition, plasma GDF15, as measured by ELISA, correlated with symptom severity for CMT1A. However, in the severely affected individuals with PMP22 exon 4 deletion or p.His12Pro, these biomarkers were within the range of variability of CMT1A and controls, although they had more pronounced nerve hypertrophy. This study adds p.His12Pro and confirms PMP22 exon 4 deletion as causes of severe CMT, whereas the previously unknown splice variant p.Glu60Lys leads to mild axonal neuropathy. Our results suggest that GDF15 and NFL do not distinguish CMT1A from advanced hypertrophic neuropathy caused by rare PMP22 variants.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":" ","pages":"291-301"},"PeriodicalIF":1.6000,"publicationDate":"2023-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10545620/pdf/","citationCount":"0","resultStr":"{\"title\":\"Rare PMP22 variants in mild to severe neuropathy uncorrelated to plasma GDF15 or neurofilament light.\",\"authors\":\"Edouard Palu, Julius Järvilehto, Jana Pennonen, Nadine Huber, Sanna-Kaisa Herukka, Annakaisa Haapasalo, Pirjo Isohanni, Henna Tyynismaa, Mari Auranen, Emil Ylikallio\",\"doi\":\"10.1007/s10048-023-00729-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Charcot-Marie-Tooth disease (CMT) is a heterogeneous set of hereditary neuropathies whose genetic causes are not fully understood. Here, we characterize three previously unknown variants in PMP22 and assess their effect on the recently described potential CMT biomarkers' growth differentiation factor 15 (GDF15) and neurofilament light (NFL): first, a heterozygous PMP22 c.178G > A (p.Glu60Lys) in one mother-son pair with adult-onset mild axonal neuropathy. The variant led to abnormal splicing, confirmed in fibroblasts by reverse transcription PCR. Second, a de novo PMP22 c.35A > C (p.His12Pro), and third, a heterozygous 3.2 kb deletion predicting loss of exon 4. The latter two had severe CMT and ultrasonography showing strong nerve enlargement similar to a previous case of exon 4 loss due to a larger deletion. We further studied patients with PMP22 duplication (CMT1A) finding slightly elevated plasma NFL, as measured by the single molecule array immunoassay (SIMOA). In addition, plasma GDF15, as measured by ELISA, correlated with symptom severity for CMT1A. However, in the severely affected individuals with PMP22 exon 4 deletion or p.His12Pro, these biomarkers were within the range of variability of CMT1A and controls, although they had more pronounced nerve hypertrophy. This study adds p.His12Pro and confirms PMP22 exon 4 deletion as causes of severe CMT, whereas the previously unknown splice variant p.Glu60Lys leads to mild axonal neuropathy. Our results suggest that GDF15 and NFL do not distinguish CMT1A from advanced hypertrophic neuropathy caused by rare PMP22 variants.</p>\",\"PeriodicalId\":56106,\"journal\":{\"name\":\"Neurogenetics\",\"volume\":\" \",\"pages\":\"291-301\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2023-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10545620/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurogenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10048-023-00729-5\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/8/22 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00729-5","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/8/22 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Rare PMP22 variants in mild to severe neuropathy uncorrelated to plasma GDF15 or neurofilament light.
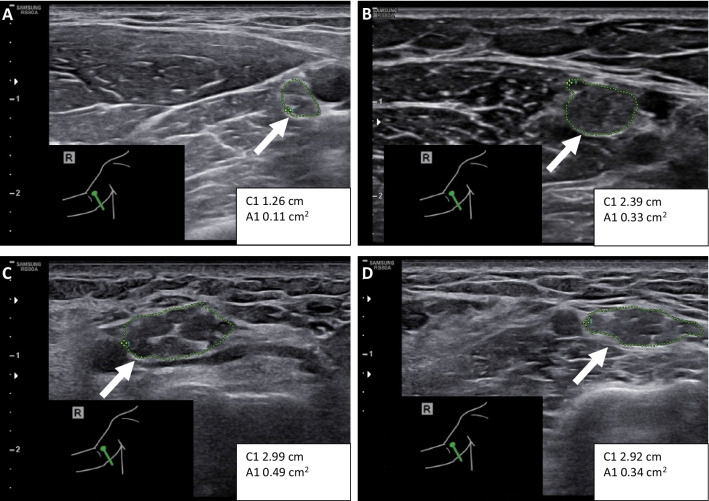
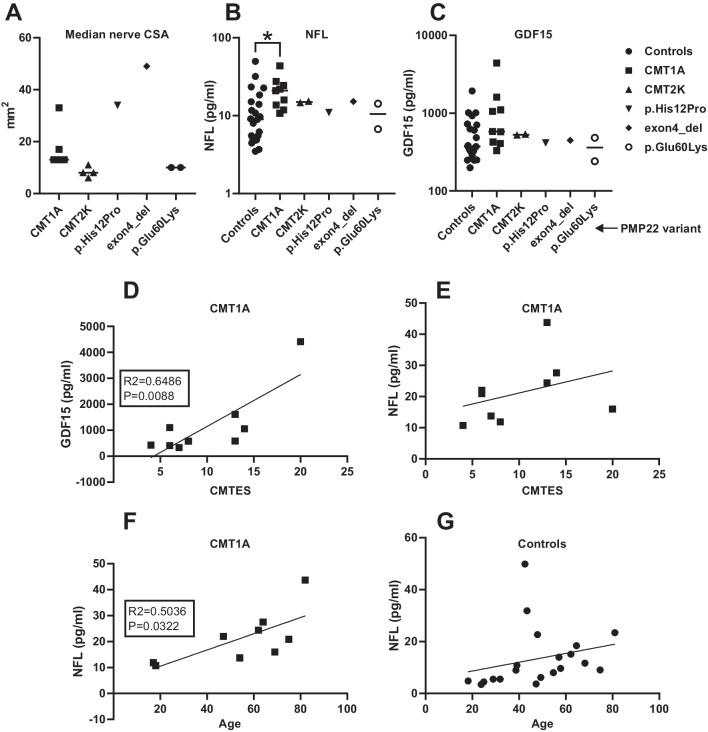
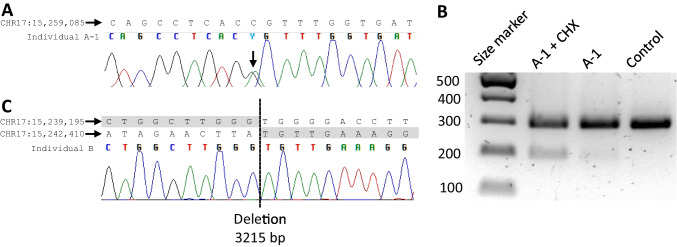
Charcot-Marie-Tooth disease (CMT) is a heterogeneous set of hereditary neuropathies whose genetic causes are not fully understood. Here, we characterize three previously unknown variants in PMP22 and assess their effect on the recently described potential CMT biomarkers' growth differentiation factor 15 (GDF15) and neurofilament light (NFL): first, a heterozygous PMP22 c.178G > A (p.Glu60Lys) in one mother-son pair with adult-onset mild axonal neuropathy. The variant led to abnormal splicing, confirmed in fibroblasts by reverse transcription PCR. Second, a de novo PMP22 c.35A > C (p.His12Pro), and third, a heterozygous 3.2 kb deletion predicting loss of exon 4. The latter two had severe CMT and ultrasonography showing strong nerve enlargement similar to a previous case of exon 4 loss due to a larger deletion. We further studied patients with PMP22 duplication (CMT1A) finding slightly elevated plasma NFL, as measured by the single molecule array immunoassay (SIMOA). In addition, plasma GDF15, as measured by ELISA, correlated with symptom severity for CMT1A. However, in the severely affected individuals with PMP22 exon 4 deletion or p.His12Pro, these biomarkers were within the range of variability of CMT1A and controls, although they had more pronounced nerve hypertrophy. This study adds p.His12Pro and confirms PMP22 exon 4 deletion as causes of severe CMT, whereas the previously unknown splice variant p.Glu60Lys leads to mild axonal neuropathy. Our results suggest that GDF15 and NFL do not distinguish CMT1A from advanced hypertrophic neuropathy caused by rare PMP22 variants.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


