8名典型和非典型重症联合免疫缺陷患者的临床、免疫学和分子研究结果:通过全外显子组测序鉴定7个新型突变基因
IF 5
3区 医学
Q1 GENETICS & HEREDITY
引用次数: 0
摘要
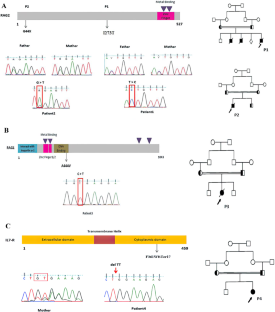
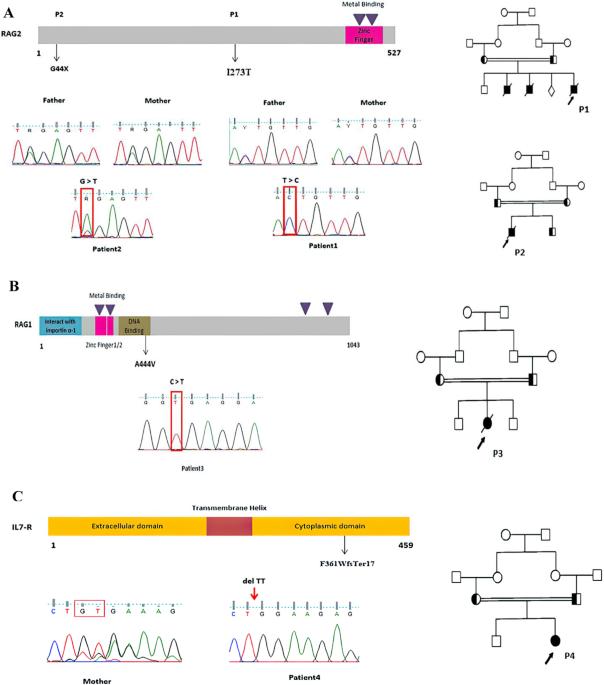
严重联合免疫缺陷症(SCID)是与危及生命的感染有关的严重先天性免疫系统错误之一。SCID 表型的变化,尤其是非典型性 SCID,可能会导致诊断的严重延误。因此,SCID 患者需要接受早期诊断。在此,我们描述了四名 SCID 和非典型 SCID 患者的临床表现和遗传结果。所有患者(4 男 4 女)均在出生后 6 个月内出现 SCID 表型。突变基因包括 RAG2(p.I273T,p.G44X)、IL7R(p.F361WfsTer17)、ADA(c.780+1G>A)、JAK3(p.Q228Ter)、LIG4(p.G428R)和 LAT(p.Y207fsTer33),以及之前报道的 RAG1(p.A444V)的错义突变。本研究是第二次报道 SCID 患者的 LAT 缺乏症。此外,所有变异均通过桑格测序在患者及其父母中证实为杂合状态。我们的研究结果扩展了与 SCID 和漏型 SCID 表型相关的临床和分子谱,为患者的临床治疗提供了有价值的信息。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Clinical, immunological and molecular findings of 8 patients with typical and atypical severe combined immunodeficiency: identification of 7 novel mutations by whole exome sequencing
Severe combined immunodeficiency (SCID) is one of the severe inborn errors of the immune system associated with life-threatening infections. Variations in SCID phenotypes, especially atypical SCID, may cause a significant delay in diagnosis. Therefore, SCID patients need to receive an early diagnosis. Here, we describe the clinical manifestations and genetic results of four SCID and atypical SCID patients. All patients (4 males and 4 females) in early infancy presented with SCID phenotypes within 6 months of birth. The mutations include RAG2 (p.I273T,p.G44X), IL7R (p.F361WfsTer17), ADA (c.780+1G>A), JAK3 (p.Q228Ter), LIG4 (p.G428R), and LAT (p.Y207fsTer33), as well as a previously reported missense mutation in RAG1 (p.A444V). The second report of LAT deficiency in SCID patients is presented in this study. Moreover, all variants were confirmed in patients and their parents as a heterozygous state by Sanger sequencing. The results of our study expand the clinical and molecular spectrum associated with SCID and leaky SCID phenotypes and provide valuable information for the clinical management of the patients.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Genes and immunity
医学-免疫学
CiteScore
8.90
自引率
4.00%
发文量
28
审稿时长
6-12 weeks
期刊介绍:
Genes & Immunity emphasizes studies investigating how genetic, genomic and functional variations affect immune cells and the immune system, and associated processes in the regulation of health and disease. It further highlights articles on the transcriptional and posttranslational control of gene products involved in signaling pathways regulating immune cells, and protective and destructive immune responses.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


