{"title":"用于疾病预测的基因水平甲基化鉴定。","authors":"Jisha Augustine, A S Jereesh","doi":"10.1007/s12539-023-00584-w","DOIUrl":null,"url":null,"abstract":"<p><p>DNA methylation is an epigenetic alteration that plays a fundamental part in governing gene regulatory processes. The DNA methylation mechanism affixes methyl groups to distinct cytosine residues, influencing chromatin architectures. Multiple studies have demonstrated that DNA methylation's regulatory effect on genes is linked to the beginning and progression of several disorders. Researchers have recently uncovered thousands of phenotype-related methylation sites through the epigenome-wide association study (EWAS). However, combining the methylation levels of several sites within a gene and determining the gene-level DNA methylation remains challenging. In this study, we proposed the supervised UMAP Assisted Gene-level Methylation method (sUAGM) for disease prediction based on supervised UMAP (Uniform Manifold Approximation and Projection), a manifold learning-based method for reducing dimensionality. The methylation values at the gene level generated using the proposed method are evaluated by employing various feature selection and classification algorithms on three distinct DNA methylation datasets derived from blood samples. The performance has been assessed employing classification accuracy, F-1 score, Mathews Correlation Coefficient (MCC), Kappa, Classification Success Index (CSI) and Jaccard Index. The Support Vector Machine with the linear kernel (SVML) classifier with Recursive Feature Elimination (RFE) performs best across all three datasets. From comparative analysis, our method outperformed existing gene-level and site-level approaches by achieving 100% accuracy and F1-score with fewer genes. The functional analysis of the top 28 genes selected from the Parkinson's disease dataset revealed a significant association with the disease.</p>","PeriodicalId":13670,"journal":{"name":"Interdisciplinary Sciences: Computational Life Sciences","volume":" ","pages":"678-695"},"PeriodicalIF":3.9000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Identification of gene-level methylation for disease prediction.\",\"authors\":\"Jisha Augustine, A S Jereesh\",\"doi\":\"10.1007/s12539-023-00584-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>DNA methylation is an epigenetic alteration that plays a fundamental part in governing gene regulatory processes. The DNA methylation mechanism affixes methyl groups to distinct cytosine residues, influencing chromatin architectures. Multiple studies have demonstrated that DNA methylation's regulatory effect on genes is linked to the beginning and progression of several disorders. Researchers have recently uncovered thousands of phenotype-related methylation sites through the epigenome-wide association study (EWAS). However, combining the methylation levels of several sites within a gene and determining the gene-level DNA methylation remains challenging. In this study, we proposed the supervised UMAP Assisted Gene-level Methylation method (sUAGM) for disease prediction based on supervised UMAP (Uniform Manifold Approximation and Projection), a manifold learning-based method for reducing dimensionality. The methylation values at the gene level generated using the proposed method are evaluated by employing various feature selection and classification algorithms on three distinct DNA methylation datasets derived from blood samples. The performance has been assessed employing classification accuracy, F-1 score, Mathews Correlation Coefficient (MCC), Kappa, Classification Success Index (CSI) and Jaccard Index. The Support Vector Machine with the linear kernel (SVML) classifier with Recursive Feature Elimination (RFE) performs best across all three datasets. From comparative analysis, our method outperformed existing gene-level and site-level approaches by achieving 100% accuracy and F1-score with fewer genes. The functional analysis of the top 28 genes selected from the Parkinson's disease dataset revealed a significant association with the disease.</p>\",\"PeriodicalId\":13670,\"journal\":{\"name\":\"Interdisciplinary Sciences: Computational Life Sciences\",\"volume\":\" \",\"pages\":\"678-695\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2023-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Interdisciplinary Sciences: Computational Life Sciences\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s12539-023-00584-w\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/8/21 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Interdisciplinary Sciences: Computational Life Sciences","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s12539-023-00584-w","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/8/21 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Identification of gene-level methylation for disease prediction.
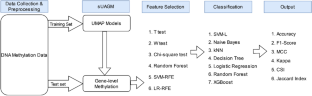
DNA methylation is an epigenetic alteration that plays a fundamental part in governing gene regulatory processes. The DNA methylation mechanism affixes methyl groups to distinct cytosine residues, influencing chromatin architectures. Multiple studies have demonstrated that DNA methylation's regulatory effect on genes is linked to the beginning and progression of several disorders. Researchers have recently uncovered thousands of phenotype-related methylation sites through the epigenome-wide association study (EWAS). However, combining the methylation levels of several sites within a gene and determining the gene-level DNA methylation remains challenging. In this study, we proposed the supervised UMAP Assisted Gene-level Methylation method (sUAGM) for disease prediction based on supervised UMAP (Uniform Manifold Approximation and Projection), a manifold learning-based method for reducing dimensionality. The methylation values at the gene level generated using the proposed method are evaluated by employing various feature selection and classification algorithms on three distinct DNA methylation datasets derived from blood samples. The performance has been assessed employing classification accuracy, F-1 score, Mathews Correlation Coefficient (MCC), Kappa, Classification Success Index (CSI) and Jaccard Index. The Support Vector Machine with the linear kernel (SVML) classifier with Recursive Feature Elimination (RFE) performs best across all three datasets. From comparative analysis, our method outperformed existing gene-level and site-level approaches by achieving 100% accuracy and F1-score with fewer genes. The functional analysis of the top 28 genes selected from the Parkinson's disease dataset revealed a significant association with the disease.
期刊介绍:
Interdisciplinary Sciences--Computational Life Sciences aims to cover the most recent and outstanding developments in interdisciplinary areas of sciences, especially focusing on computational life sciences, an area that is enjoying rapid development at the forefront of scientific research and technology.
The journal publishes original papers of significant general interest covering recent research and developments. Articles will be published rapidly by taking full advantage of internet technology for online submission and peer-reviewing of manuscripts, and then by publishing OnlineFirstTM through SpringerLink even before the issue is built or sent to the printer.
The editorial board consists of many leading scientists with international reputation, among others, Luc Montagnier (UNESCO, France), Dennis Salahub (University of Calgary, Canada), Weitao Yang (Duke University, USA). Prof. Dongqing Wei at the Shanghai Jiatong University is appointed as the editor-in-chief; he made important contributions in bioinformatics and computational physics and is best known for his ground-breaking works on the theory of ferroelectric liquids. With the help from a team of associate editors and the editorial board, an international journal with sound reputation shall be created.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


