Rita Sianga-Mete, Penelope Hartnady, Wimbai Caroline Mandikumba, Kayleigh Rutherford, Christopher Brian Currin, Florence Phelanyane, Sabina Stefan, Sergei L Kosakovsky Pond, Darren Patrick Martin
{"title":"病毒基因组序列数据集显示了链特异性取代偏差的普遍证据,这些证据最好使用不可逆核苷酸取代模型来描述。","authors":"Rita Sianga-Mete, Penelope Hartnady, Wimbai Caroline Mandikumba, Kayleigh Rutherford, Christopher Brian Currin, Florence Phelanyane, Sabina Stefan, Sergei L Kosakovsky Pond, Darren Patrick Martin","doi":"10.21203/rs.3.rs-2407778/v2","DOIUrl":null,"url":null,"abstract":"<p><p>Most phylogenetic trees are inferred using time-reversible evolutionary models that assume that the relative rates of substitution for any given pair of nucleotides are the same regardless of the direction of the substitutions. However, there is no reason to assume that the underlying biochemical mutational processes that cause substitutions are similarly symmetrical. We consider two non-reversible nucleotide substitution models: (1) a 6-rate non-reversible model (NREV6) that is applicable to analyzing mutational processes in double-stranded genomes in that complementary substitutions occur at identical rates; and (2) a 12-rate non-reversible model (NREV12) that is applicable to analyzing mutational processes in single-stranded (ss) genomes in that all substitution types are free to occur at different rates. Using likelihood ratio and Akaike Information Criterion-based model tests, we show that, surprisingly, NREV12 provided a significantly better fit than the General Time Reversible (GTR) and NREV6 models to 21/31 dsRNA and 20/30 dsDNA datasets. As expected, however, NREV12 provided a significantly better fit to 24/33 ssDNA and 40/47 ssRNA datasets. We tested how non-reversibility impacts the accuracy with which phylogenetic trees are inferred. As simulated degrees of non-reversibility (DNR) increased, the tree topology inferences using both NREV12 and GTR became more accurate, whereas inferred tree branch lengths became less accurate. We conclude that while non-reversible models should be helpful in the analysis of mutational processes in most virus species, there is no pressing need to use these models for routine phylogenetic inference.</p>","PeriodicalId":21039,"journal":{"name":"Research Square","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2025-08-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9810213/pdf/","citationCount":"0","resultStr":"{\"title\":\"Viral genome sequence datasets display pervasive evidence of strand-specific substitution biases that are best described using non-reversible nucleotide substitution models.\",\"authors\":\"Rita Sianga-Mete, Penelope Hartnady, Wimbai Caroline Mandikumba, Kayleigh Rutherford, Christopher Brian Currin, Florence Phelanyane, Sabina Stefan, Sergei L Kosakovsky Pond, Darren Patrick Martin\",\"doi\":\"10.21203/rs.3.rs-2407778/v2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Most phylogenetic trees are inferred using time-reversible evolutionary models that assume that the relative rates of substitution for any given pair of nucleotides are the same regardless of the direction of the substitutions. However, there is no reason to assume that the underlying biochemical mutational processes that cause substitutions are similarly symmetrical. We consider two non-reversible nucleotide substitution models: (1) a 6-rate non-reversible model (NREV6) that is applicable to analyzing mutational processes in double-stranded genomes in that complementary substitutions occur at identical rates; and (2) a 12-rate non-reversible model (NREV12) that is applicable to analyzing mutational processes in single-stranded (ss) genomes in that all substitution types are free to occur at different rates. Using likelihood ratio and Akaike Information Criterion-based model tests, we show that, surprisingly, NREV12 provided a significantly better fit than the General Time Reversible (GTR) and NREV6 models to 21/31 dsRNA and 20/30 dsDNA datasets. As expected, however, NREV12 provided a significantly better fit to 24/33 ssDNA and 40/47 ssRNA datasets. We tested how non-reversibility impacts the accuracy with which phylogenetic trees are inferred. As simulated degrees of non-reversibility (DNR) increased, the tree topology inferences using both NREV12 and GTR became more accurate, whereas inferred tree branch lengths became less accurate. We conclude that while non-reversible models should be helpful in the analysis of mutational processes in most virus species, there is no pressing need to use these models for routine phylogenetic inference.</p>\",\"PeriodicalId\":21039,\"journal\":{\"name\":\"Research Square\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-08-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9810213/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Research Square\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.21203/rs.3.rs-2407778/v2\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Research Square","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.21203/rs.3.rs-2407778/v2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Viral genome sequence datasets display pervasive evidence of strand-specific substitution biases that are best described using non-reversible nucleotide substitution models.
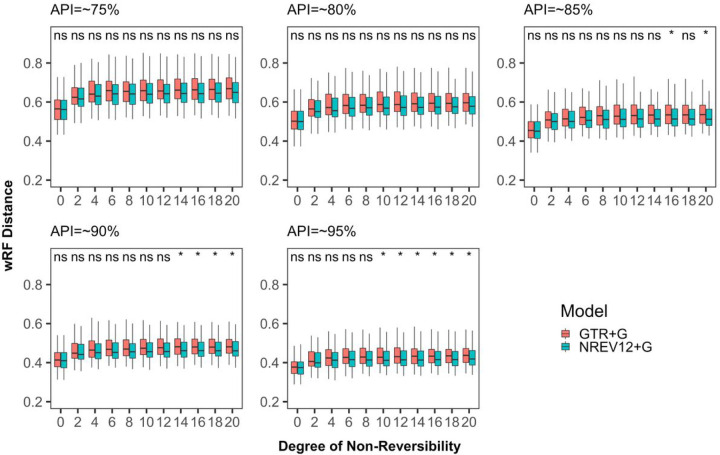
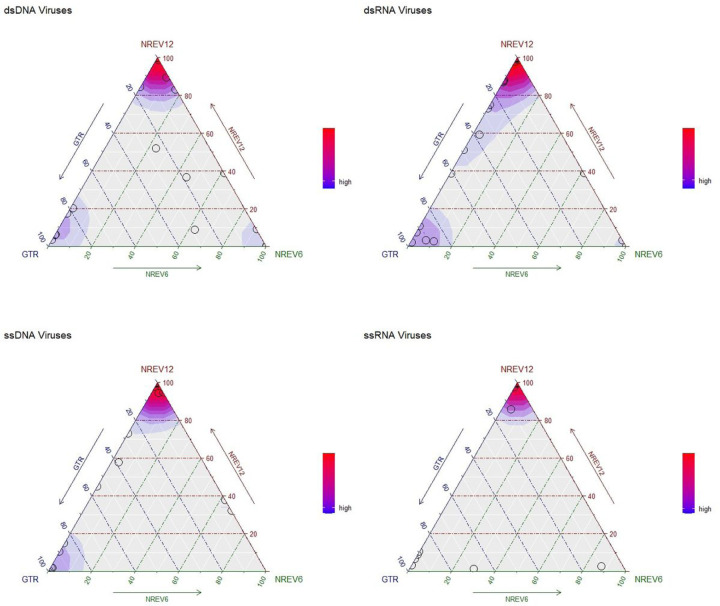
Most phylogenetic trees are inferred using time-reversible evolutionary models that assume that the relative rates of substitution for any given pair of nucleotides are the same regardless of the direction of the substitutions. However, there is no reason to assume that the underlying biochemical mutational processes that cause substitutions are similarly symmetrical. We consider two non-reversible nucleotide substitution models: (1) a 6-rate non-reversible model (NREV6) that is applicable to analyzing mutational processes in double-stranded genomes in that complementary substitutions occur at identical rates; and (2) a 12-rate non-reversible model (NREV12) that is applicable to analyzing mutational processes in single-stranded (ss) genomes in that all substitution types are free to occur at different rates. Using likelihood ratio and Akaike Information Criterion-based model tests, we show that, surprisingly, NREV12 provided a significantly better fit than the General Time Reversible (GTR) and NREV6 models to 21/31 dsRNA and 20/30 dsDNA datasets. As expected, however, NREV12 provided a significantly better fit to 24/33 ssDNA and 40/47 ssRNA datasets. We tested how non-reversibility impacts the accuracy with which phylogenetic trees are inferred. As simulated degrees of non-reversibility (DNR) increased, the tree topology inferences using both NREV12 and GTR became more accurate, whereas inferred tree branch lengths became less accurate. We conclude that while non-reversible models should be helpful in the analysis of mutational processes in most virus species, there is no pressing need to use these models for routine phylogenetic inference.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


