Combined approaches for increasing fetal hemoglobin (HbF) and de novo production of adult hemoglobin (HbA) in erythroid cells from β-thalassemia patients: treatment with HbF inducers and CRISPR-Cas9 based genome editing.
{"title":"Combined approaches for increasing fetal hemoglobin (HbF) and <i>de novo</i> production of adult hemoglobin (HbA) in erythroid cells from β-thalassemia patients: treatment with HbF inducers and CRISPR-Cas9 based genome editing.","authors":"Alessia Finotti, Roberto Gambari","doi":"10.3389/fgeed.2023.1204536","DOIUrl":null,"url":null,"abstract":"<p><p>Genome editing (GE) is one of the most efficient and useful molecular approaches to correct the effects of gene mutations in hereditary monogenetic diseases, including β-thalassemia. CRISPR-Cas9 gene editing has been proposed for effective correction of the β-thalassemia mutation, obtaining high-level \"<i>de novo</i>\" production of adult hemoglobin (HbA). In addition to the correction of the primary gene mutations causing β-thalassemia, several reports demonstrate that gene editing can be employed to increase fetal hemoglobin (HbF), obtaining important clinical benefits in treated β-thalassemia patients. This important objective can be achieved through CRISPR-Cas9 disruption of genes encoding transcriptional repressors of γ-globin gene expression (such as <i>BCL11A, SOX6, KLF-1</i>) or their binding sites in the HBG promoter, mimicking non-deletional and deletional HPFH mutations. These two approaches (β-globin gene correction and genome editing of the genes encoding repressors of γ-globin gene transcription) can be, at least in theory, combined. However, since multiplex CRISPR-Cas9 gene editing is associated with documented evidence concerning possible genotoxicity, this review is focused on the possibility to combine pharmacologically-mediated HbF induction protocols with the \"<i>de novo</i>\" production of HbA using CRISPR-Cas9 gene editing.</p>","PeriodicalId":73086,"journal":{"name":"Frontiers in genome editing","volume":"5 ","pages":"1204536"},"PeriodicalIF":4.9000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10387548/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in genome editing","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fgeed.2023.1204536","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
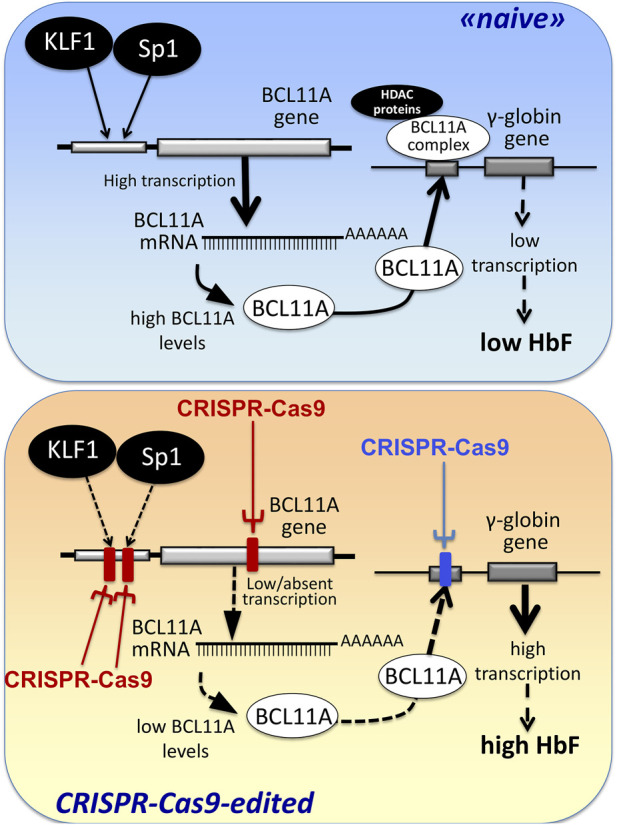
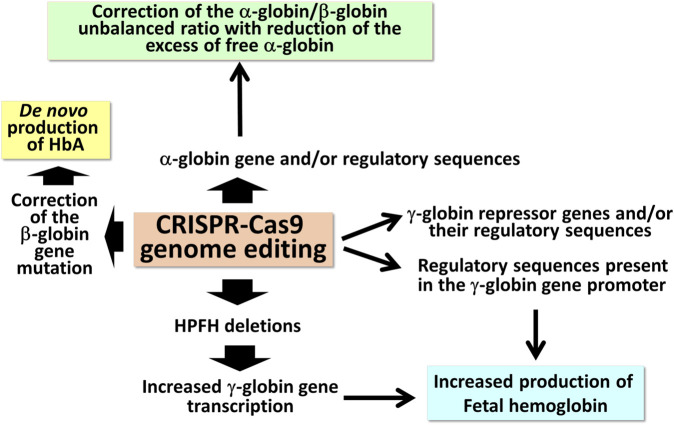
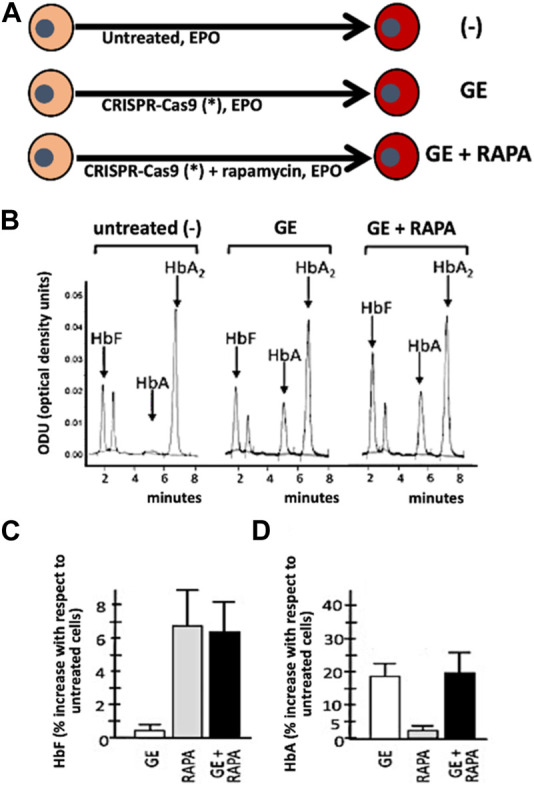
Genome editing (GE) is one of the most efficient and useful molecular approaches to correct the effects of gene mutations in hereditary monogenetic diseases, including β-thalassemia. CRISPR-Cas9 gene editing has been proposed for effective correction of the β-thalassemia mutation, obtaining high-level "de novo" production of adult hemoglobin (HbA). In addition to the correction of the primary gene mutations causing β-thalassemia, several reports demonstrate that gene editing can be employed to increase fetal hemoglobin (HbF), obtaining important clinical benefits in treated β-thalassemia patients. This important objective can be achieved through CRISPR-Cas9 disruption of genes encoding transcriptional repressors of γ-globin gene expression (such as BCL11A, SOX6, KLF-1) or their binding sites in the HBG promoter, mimicking non-deletional and deletional HPFH mutations. These two approaches (β-globin gene correction and genome editing of the genes encoding repressors of γ-globin gene transcription) can be, at least in theory, combined. However, since multiplex CRISPR-Cas9 gene editing is associated with documented evidence concerning possible genotoxicity, this review is focused on the possibility to combine pharmacologically-mediated HbF induction protocols with the "de novo" production of HbA using CRISPR-Cas9 gene editing.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


