{"title":"Metformin Induces Lipogenesis and Apoptosis in H4IIE Hepatocellular Carcinoma Cells.","authors":"Deokbae Park, Sookyoung Lee, Hyejin Boo","doi":"10.12717/DR.2023.27.2.77","DOIUrl":null,"url":null,"abstract":"<p><p>Metformin is the most widely used anti-diabetic drug that helps maintain normal blood glucose levels primarily by suppressing hepatic gluconeogenesis in type II diabetic patients. We previously found that metformin induces apoptotic death in H4IIE rat hepatocellular carcinoma cells. Despite its anti-diabetic roles, the effect of metformin on hepatic de novo lipogenesis (DNL) remains unclear. We investigated the effect of metformin on hepatic DNL and apoptotic cell death in H4IIE cells. Metformin treatment stimulated glucose consumption, lactate production, intracellular fat accumulation, and the expressions of lipogenic proteins. It also stimulated apoptosis but reduced autophagic responses. These metformin-induced changes were clearly reversed by compound C, an inhibitor of AMP-activated protein kinase (AMPK). Interestingly, metformin massively increased the production of reactive oxygen species (ROS), which was completely blocked by compound C. Metformin also stimulated the phosphorylation of p38 mitogen-activated protein kinase (p38MAPK). Finally, inhibition of p38MAPK mimicked the effects of compound C, and suppressed the metformin-induced fat accumulation and apoptosis. Taken together, metformin stimulates dysregulated glucose metabolism, intracellular fat accumulation, and apoptosis. Our findings suggest that metformin induces excessive glucose-induced DNL, oxidative stress by ROS generation, activation of AMPK and p38MAPK, suppression of autophagy, and ultimately apoptosis.</p>","PeriodicalId":72791,"journal":{"name":"Development & reproduction","volume":"27 2","pages":"77-89"},"PeriodicalIF":0.0000,"publicationDate":"2023-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/57/77/dr-27-2-77.PMC10390098.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Development & reproduction","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.12717/DR.2023.27.2.77","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/6/30 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
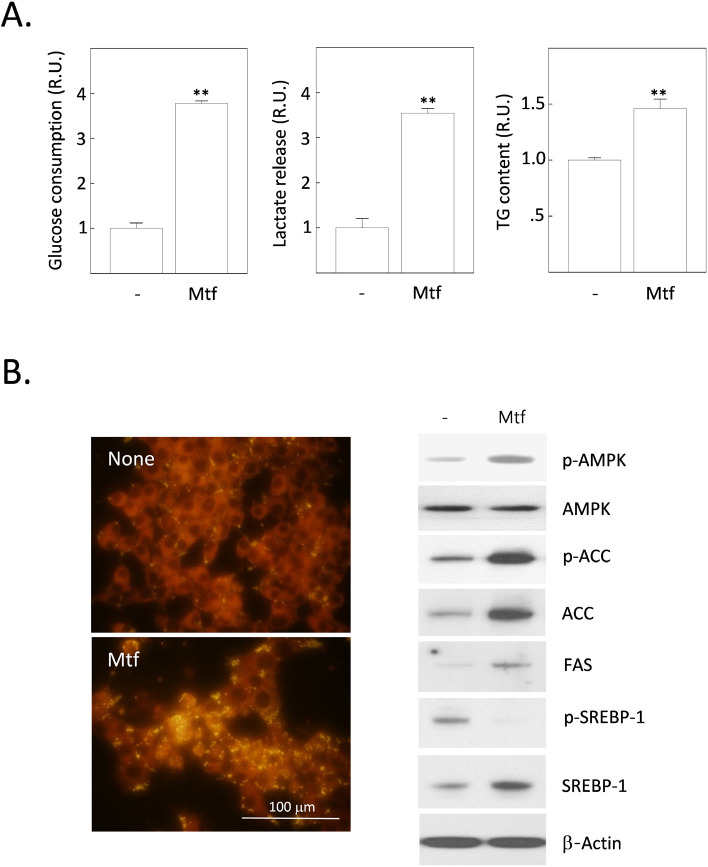
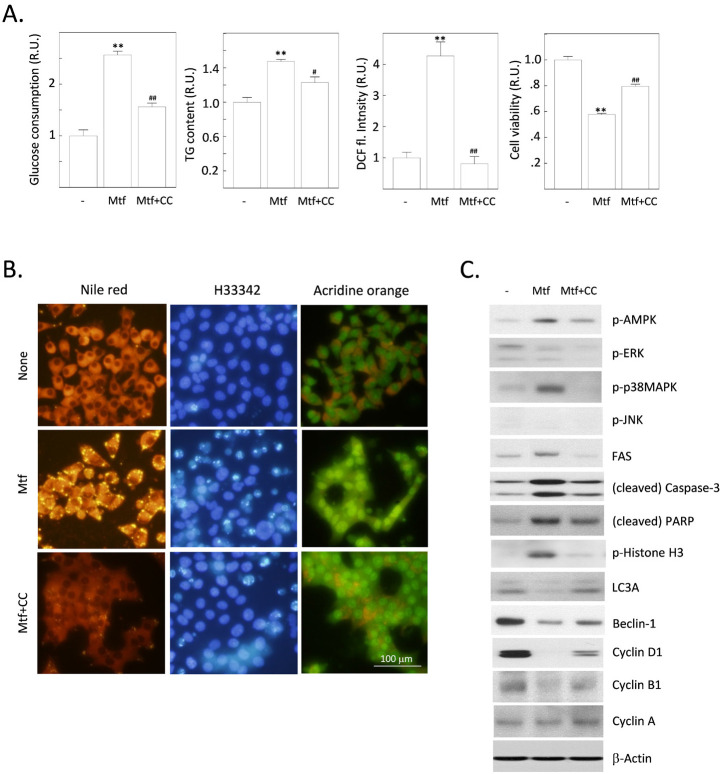
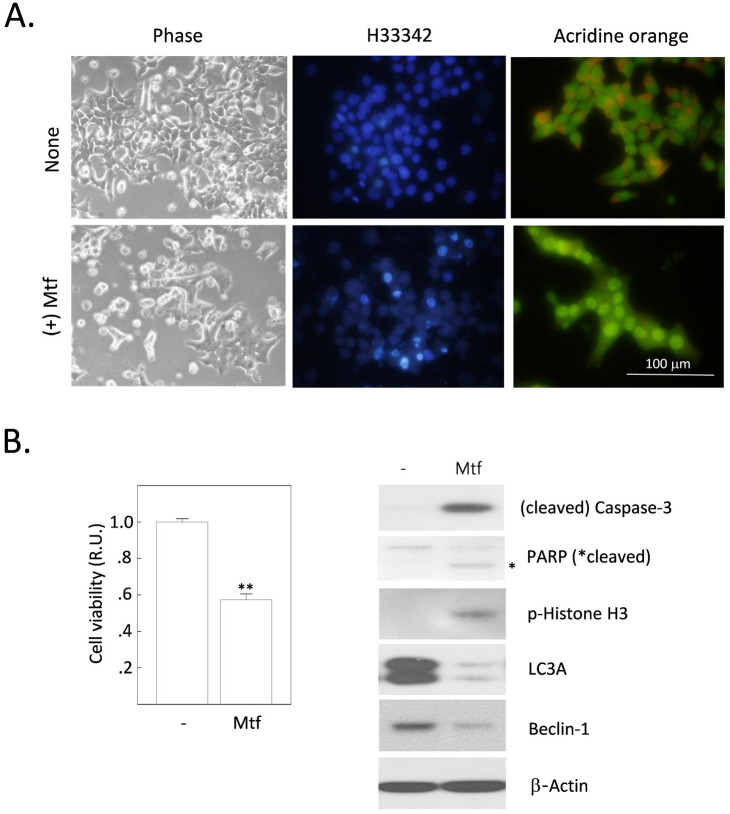
Metformin is the most widely used anti-diabetic drug that helps maintain normal blood glucose levels primarily by suppressing hepatic gluconeogenesis in type II diabetic patients. We previously found that metformin induces apoptotic death in H4IIE rat hepatocellular carcinoma cells. Despite its anti-diabetic roles, the effect of metformin on hepatic de novo lipogenesis (DNL) remains unclear. We investigated the effect of metformin on hepatic DNL and apoptotic cell death in H4IIE cells. Metformin treatment stimulated glucose consumption, lactate production, intracellular fat accumulation, and the expressions of lipogenic proteins. It also stimulated apoptosis but reduced autophagic responses. These metformin-induced changes were clearly reversed by compound C, an inhibitor of AMP-activated protein kinase (AMPK). Interestingly, metformin massively increased the production of reactive oxygen species (ROS), which was completely blocked by compound C. Metformin also stimulated the phosphorylation of p38 mitogen-activated protein kinase (p38MAPK). Finally, inhibition of p38MAPK mimicked the effects of compound C, and suppressed the metformin-induced fat accumulation and apoptosis. Taken together, metformin stimulates dysregulated glucose metabolism, intracellular fat accumulation, and apoptosis. Our findings suggest that metformin induces excessive glucose-induced DNL, oxidative stress by ROS generation, activation of AMPK and p38MAPK, suppression of autophagy, and ultimately apoptosis.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


