Wenbo Yu*, David J. Weber and Alexander D. MacKerell Jr.*,
{"title":"Integrated Covalent Drug Design Workflow Using Site Identification by Ligand Competitive Saturation","authors":"Wenbo Yu*, David J. Weber and Alexander D. MacKerell Jr.*, ","doi":"10.1021/acs.jctc.3c00232","DOIUrl":null,"url":null,"abstract":"<p >Covalent drug design is an important component in drug discovery. Traditional drugs interact with their target in a reversible equilibrium, while irreversible covalent drugs increase the drug–target interaction duration by forming a covalent bond with targeted residues and thus may offer a more effective therapeutic approach. To facilitate the design of this class of ligands, computational methods can be used to help identify reactive nucleophilic residues, frequently cysteines, on a target protein for covalent binding, to test various warhead groups for their potential reactivities, and to predict noncovalent contributions to binding that can facilitate drug–target interactions that are important for binding specificity. To further aid covalent drug design, we extended a functional group mapping approach based on explicit solvent all-atom molecular simulations (SILCS: site identification by ligand competitive saturation) that intrinsically considers protein flexibility, functional group, and protein desolvation along with functional group–protein interactions. Through docking of a library of representative warhead fragments using SILCS-Monte Carlo (SILCS-MC), reactive cysteines can be correctly identified for proteins being tested. Furthermore, a machine learning model was trained to quantify the effectiveness of various warhead groups for proteins using metrics from SILCS-MC as well as experimental model compound warhead reactivity data. The ability to rank covalent molecular binders with similar warheads using SILCS ligand grid free energy (LGFE) ranking was also tested for several proteins. Based on these tools, an integrated SILCS-based workflow was developed, named SILCS-Covalent, which can both qualitatively and quantitatively inform covalent drug discovery.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"19 10","pages":"3007–3021"},"PeriodicalIF":5.5000,"publicationDate":"2023-04-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.3c00232","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 1
Abstract
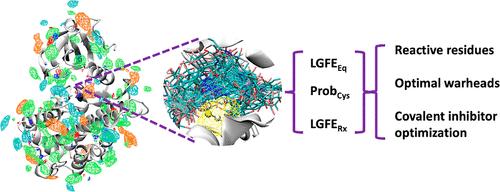
Covalent drug design is an important component in drug discovery. Traditional drugs interact with their target in a reversible equilibrium, while irreversible covalent drugs increase the drug–target interaction duration by forming a covalent bond with targeted residues and thus may offer a more effective therapeutic approach. To facilitate the design of this class of ligands, computational methods can be used to help identify reactive nucleophilic residues, frequently cysteines, on a target protein for covalent binding, to test various warhead groups for their potential reactivities, and to predict noncovalent contributions to binding that can facilitate drug–target interactions that are important for binding specificity. To further aid covalent drug design, we extended a functional group mapping approach based on explicit solvent all-atom molecular simulations (SILCS: site identification by ligand competitive saturation) that intrinsically considers protein flexibility, functional group, and protein desolvation along with functional group–protein interactions. Through docking of a library of representative warhead fragments using SILCS-Monte Carlo (SILCS-MC), reactive cysteines can be correctly identified for proteins being tested. Furthermore, a machine learning model was trained to quantify the effectiveness of various warhead groups for proteins using metrics from SILCS-MC as well as experimental model compound warhead reactivity data. The ability to rank covalent molecular binders with similar warheads using SILCS ligand grid free energy (LGFE) ranking was also tested for several proteins. Based on these tools, an integrated SILCS-based workflow was developed, named SILCS-Covalent, which can both qualitatively and quantitatively inform covalent drug discovery.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


