{"title":"Identification of an 85-kb Heterozygous 4p Microdeletion With Full Genome Analysis in Autosomal Dominant Charcot-Marie-Tooth Disease.","authors":"Hsueh Wen Hsueh, Hsiao-Jung Kao, Chi-Chao Chao, Sung-Ju Hsueh, Yu-Ning Huang, Wan-Jia Lin, Jen-Ping Su, Horng-Tzer Shy, Ti-Yen Yeh, Cheng-Chen Lin, Pui-Yan Kwok, Ni-Chung Lee, Sung-Tsang Hsieh","doi":"10.1212/NXG.0000000000200078","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong>Charcot-Marie-Tooth disease (CMT) is a syndrome of a hereditary neurodegenerative condition affecting the peripheral nervous system and is a single gene disorder. Deep phenotyping coupled with advanced genetic techniques is critical in discovering new genetic defects of rare genetic disorders such as CMT.</p><p><strong>Methods: </strong>We applied multidisciplinary investigations to examine the neurophysiology and nerve pathology in a family that fulfilled the diagnosis of CMT2. When phenotype-guided first-tier genetic tests and whole-exome sequencing did not yield a molecular diagnosis, we conducted full genome analysis by examining phased whole-genome sequencing and whole-genome optical mapping data to search for the causal variation. We then performed a systematic review to compare the reported patients with interstitial microdeletion in the short arm of chromosome 4.</p><p><strong>Results: </strong>In this family with CMT2, we reported the discovery of a heterozygous 85-kb microdeletion in the short arm of chromosome 4 (4p16.3)[NC_000004.12:g.1733926_1819031del] spanning 3 genes [<i>TACC3</i> (intron 6-exon 16), <i>FGFR3</i> (total deletion), and <i>LETM1</i> (intron 10-exon14)] that cosegregated with disease phenotypes in family members. The clinical features of peripheral nerve degeneration in our family are distinct from the well-known 4p microdeletion syndrome of Wolf-Hirschhorn syndrome, in which brain involvement is the major phenotype.</p><p><strong>Discussion: </strong>In summary, we used the full genome analysis approach to discover a new microdeletion in a family with CMT2. The deleted segment contains 3 genes (<i>TACC3</i>, <i>FGFR3</i>, and <i>LETM1</i>) that likely play a role in the pathogenesis of nerve degeneration.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"9 4","pages":"e200078"},"PeriodicalIF":3.0000,"publicationDate":"2023-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/d6/03/NXG-2023-000021.PMC10281236.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200078","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background and objectives: Charcot-Marie-Tooth disease (CMT) is a syndrome of a hereditary neurodegenerative condition affecting the peripheral nervous system and is a single gene disorder. Deep phenotyping coupled with advanced genetic techniques is critical in discovering new genetic defects of rare genetic disorders such as CMT.
Methods: We applied multidisciplinary investigations to examine the neurophysiology and nerve pathology in a family that fulfilled the diagnosis of CMT2. When phenotype-guided first-tier genetic tests and whole-exome sequencing did not yield a molecular diagnosis, we conducted full genome analysis by examining phased whole-genome sequencing and whole-genome optical mapping data to search for the causal variation. We then performed a systematic review to compare the reported patients with interstitial microdeletion in the short arm of chromosome 4.
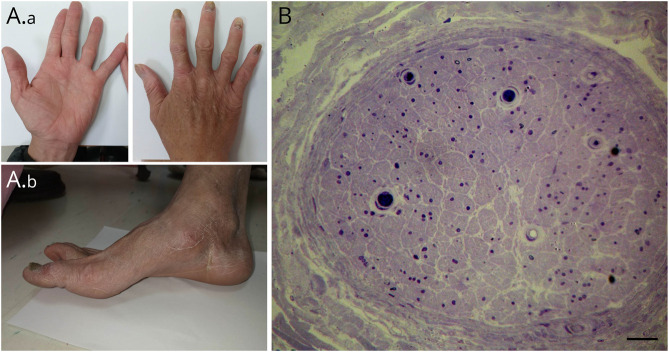
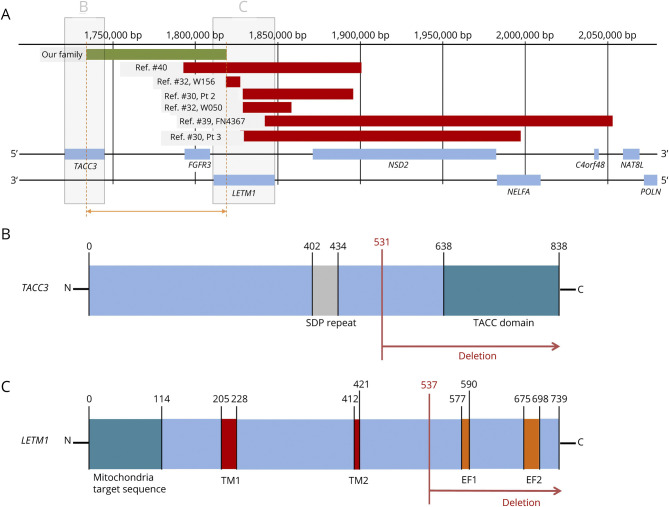
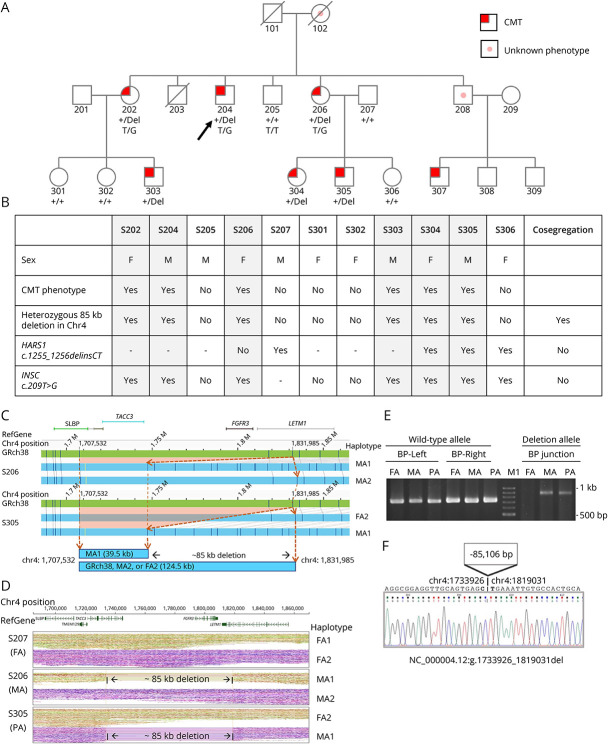
Results: In this family with CMT2, we reported the discovery of a heterozygous 85-kb microdeletion in the short arm of chromosome 4 (4p16.3)[NC_000004.12:g.1733926_1819031del] spanning 3 genes [TACC3 (intron 6-exon 16), FGFR3 (total deletion), and LETM1 (intron 10-exon14)] that cosegregated with disease phenotypes in family members. The clinical features of peripheral nerve degeneration in our family are distinct from the well-known 4p microdeletion syndrome of Wolf-Hirschhorn syndrome, in which brain involvement is the major phenotype.
Discussion: In summary, we used the full genome analysis approach to discover a new microdeletion in a family with CMT2. The deleted segment contains 3 genes (TACC3, FGFR3, and LETM1) that likely play a role in the pathogenesis of nerve degeneration.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


