David Paz , Briana E. Pinales , Barbara S. Castellanos , Isaiah Perez , Claudia B. Gil , Lourdes Jimenez Madrigal , Nayeli G. Reyes-Nava , Victoria L. Castro , Jennifer L. Sloan , Anita M. Quintana
{"title":"Abnormal chondrocyte development in a zebrafish model of cblC syndrome restored by an MMACHC cobalamin binding mutant","authors":"David Paz , Briana E. Pinales , Barbara S. Castellanos , Isaiah Perez , Claudia B. Gil , Lourdes Jimenez Madrigal , Nayeli G. Reyes-Nava , Victoria L. Castro , Jennifer L. Sloan , Anita M. Quintana","doi":"10.1016/j.diff.2023.04.003","DOIUrl":null,"url":null,"abstract":"<div><p>Variants in the <em>MMACHC</em> gene cause combined methylmalonic acidemia and homocystinuria <em>cblC</em> type, the most common inborn error of intracellular cobalamin (vitamin B12) metabolism. <em>cblC</em> is associated with neurodevelopmental, hematological, ocular, and biochemical abnormalities. In a subset of patients, mild craniofacial dysmorphia has also been described. Mouse models of <em>Mmachc</em> deletion are embryonic lethal but cause severe craniofacial phenotypes such as facial clefts. <em>MMACHC</em> encodes an enzyme required for cobalamin processing and variants in this gene result in the accumulation of two metabolites: methylmalonic acid (MMA) and homocysteine (HC). Interestingly, other inborn errors of cobalamin metabolism, such as <em>cblX</em> syndrome, are associated with mild facial phenotypes. However, the presence and severity of MMA and HC accumulation in <em>cblX</em> syndrome is not consistent with the presence or absence of facial phenotypes. Thus, the mechanisms by which mutations in <em>MMACHC</em> cause craniofacial defects are yet to be completely elucidated. Here we have characterized the craniofacial phenotypes in a zebrafish model of <em>cblC</em> (<em>hg13</em>) and performed restoration experiments with either a wildtype or a cobalamin binding deficient MMACHC protein. Homozygous mutants did not display gross morphological defects in facial development but did have abnormal chondrocyte nuclear organization and an increase in the average number of neighboring cell contacts, both phenotypes were fully penetrant. Abnormal chondrocyte nuclear organization was not associated with defects in the localization of neural crest specific markers, <em>sox10</em> (RFP transgene) or <em>barx1</em>. Both nuclear angles and the number of neighboring cell contacts were fully restored by wildtype MMACHC and a cobalamin binding deficient variant of the MMACHC protein. Collectively, these data suggest that mutation of <em>MMACHC</em> causes mild to moderate craniofacial phenotypes that are independent of cobalamin binding.</p></div>","PeriodicalId":50579,"journal":{"name":"Differentiation","volume":"131 ","pages":"Pages 74-81"},"PeriodicalIF":2.2000,"publicationDate":"2023-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Differentiation","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0301468123000245","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
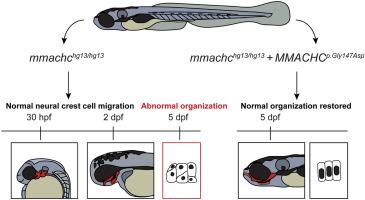
Variants in the MMACHC gene cause combined methylmalonic acidemia and homocystinuria cblC type, the most common inborn error of intracellular cobalamin (vitamin B12) metabolism. cblC is associated with neurodevelopmental, hematological, ocular, and biochemical abnormalities. In a subset of patients, mild craniofacial dysmorphia has also been described. Mouse models of Mmachc deletion are embryonic lethal but cause severe craniofacial phenotypes such as facial clefts. MMACHC encodes an enzyme required for cobalamin processing and variants in this gene result in the accumulation of two metabolites: methylmalonic acid (MMA) and homocysteine (HC). Interestingly, other inborn errors of cobalamin metabolism, such as cblX syndrome, are associated with mild facial phenotypes. However, the presence and severity of MMA and HC accumulation in cblX syndrome is not consistent with the presence or absence of facial phenotypes. Thus, the mechanisms by which mutations in MMACHC cause craniofacial defects are yet to be completely elucidated. Here we have characterized the craniofacial phenotypes in a zebrafish model of cblC (hg13) and performed restoration experiments with either a wildtype or a cobalamin binding deficient MMACHC protein. Homozygous mutants did not display gross morphological defects in facial development but did have abnormal chondrocyte nuclear organization and an increase in the average number of neighboring cell contacts, both phenotypes were fully penetrant. Abnormal chondrocyte nuclear organization was not associated with defects in the localization of neural crest specific markers, sox10 (RFP transgene) or barx1. Both nuclear angles and the number of neighboring cell contacts were fully restored by wildtype MMACHC and a cobalamin binding deficient variant of the MMACHC protein. Collectively, these data suggest that mutation of MMACHC causes mild to moderate craniofacial phenotypes that are independent of cobalamin binding.
期刊介绍:
Differentiation is a multidisciplinary journal dealing with topics relating to cell differentiation, development, cellular structure and function, and cancer. Differentiation of eukaryotes at the molecular level and the use of transgenic and targeted mutagenesis approaches to problems of differentiation are of particular interest to the journal.
The journal will publish full-length articles containing original work in any of these areas. We will also publish reviews and commentaries on topics of current interest.
The principal subject areas the journal covers are: • embryonic patterning and organogenesis
• human development and congenital malformation
• mechanisms of cell lineage commitment
• tissue homeostasis and oncogenic transformation
• establishment of cellular polarity
• stem cell differentiation
• cell reprogramming mechanisms
• stability of the differentiated state
• cell and tissue interactions in vivo and in vitro
• signal transduction pathways in development and differentiation
• carcinogenesis and cancer
• mechanisms involved in cell growth and division especially relating to cancer
• differentiation in regeneration and ageing
• therapeutic applications of differentiation processes.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


