Daitao Zhang, Xin Zhang, Bing Lyu, Yi Tian, Ying Huang, Changying Lin, Hanqiu Yan, Lei Jia, Mei Qu, Quanyi Wang
{"title":"Genomic Analysis and Antimicrobial Resistance of <i>Campylobacter jejuni</i> Isolated from Diarrheal Patients - Beijing Municipality, China, 2019-2021.","authors":"Daitao Zhang, Xin Zhang, Bing Lyu, Yi Tian, Ying Huang, Changying Lin, Hanqiu Yan, Lei Jia, Mei Qu, Quanyi Wang","doi":"10.46234/ccdcw2023.080","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong><i>Campylobacter jejuni <i>(</i>C. jejuni</i>) is the leading cause of human bacterial gastroenteritis worldwide and has a major impact on global public health. The objective of the present study was to conduct whole genome sequencing (WGS) to determine the genetic diversity, virulence factors, and determinants of antimicrobial resistance of <i>C. jejuni</i> during a 3-year surveillance period in Beijing, China.</p><p><strong>Methods: </strong>A total of 184 clinical isolates were obtained from sentinel hospital surveillance between 2019 and 2021. Antimicrobial susceptibility testing was conducted using the agar dilution method. WGS was employed to characterize the 184 <i>C. jejuni</i> strains.</p><p><strong>Results: </strong>Multilocus sequence typing analysis revealed high genetic diversity among the 184 <i>C. jejuni</i> strains, identifying 71 sequence types (STs) and 19 clonal complexes (CCs). The most prevalent ST was ST760 (6.5%), and the most common CC was CC21 (24.5%), consisting of 11 STs. High resistance rates were observed for ciprofloxacin (76.6%), nalidixic acid (76.1%), and tetracycline (71.2%). A total of 77 <i>C. jejuni</i> isolates (41.8%) exhibited multidrug resistance with 43 resistance patterns. Virulome analysis disclosed the differential distribution of virulence factors related to adherence, colonization, chemotaxis, as well as lipo-oligosaccharide and capsular polysaccharide biosynthesis. Resistome analysis demonstrated widespread resistance to quinolones and tetracycline, but low rates of macrolides resistance. The phylogeny, based on whole genome single nucleotide polymorphisms, indicated a high degree of clonality and grouped the <i>C. jejuni</i> strains into six clades. Closely related isolates that were part of a genetic cluster mostly shared a homogenous clonal complex.</p><p><strong>Conclusions: </strong>The present study emphasizes the rising resistance to quinolones and tetracycline, as well as the virulence potential and diverse genotypes identified among <i>C. jejuni</i> strains isolated from diarrheal patients in Beijing.</p>","PeriodicalId":9867,"journal":{"name":"China CDC Weekly","volume":"5 19","pages":"424-433"},"PeriodicalIF":0.0000,"publicationDate":"2023-05-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/89/04/ccdcw-5-19-424.PMC10235816.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"China CDC Weekly","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.46234/ccdcw2023.080","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Campylobacter jejuni (C. jejuni) is the leading cause of human bacterial gastroenteritis worldwide and has a major impact on global public health. The objective of the present study was to conduct whole genome sequencing (WGS) to determine the genetic diversity, virulence factors, and determinants of antimicrobial resistance of C. jejuni during a 3-year surveillance period in Beijing, China.
Methods: A total of 184 clinical isolates were obtained from sentinel hospital surveillance between 2019 and 2021. Antimicrobial susceptibility testing was conducted using the agar dilution method. WGS was employed to characterize the 184 C. jejuni strains.
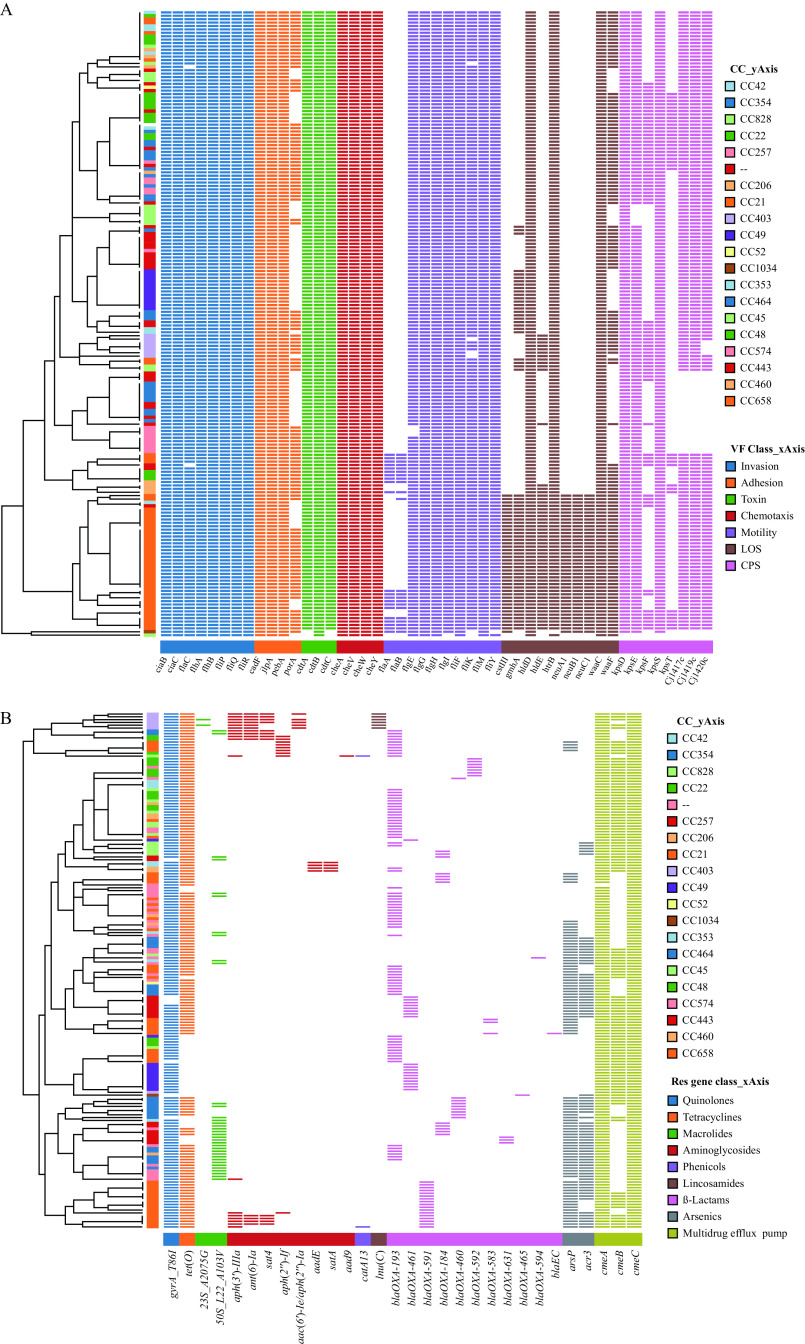
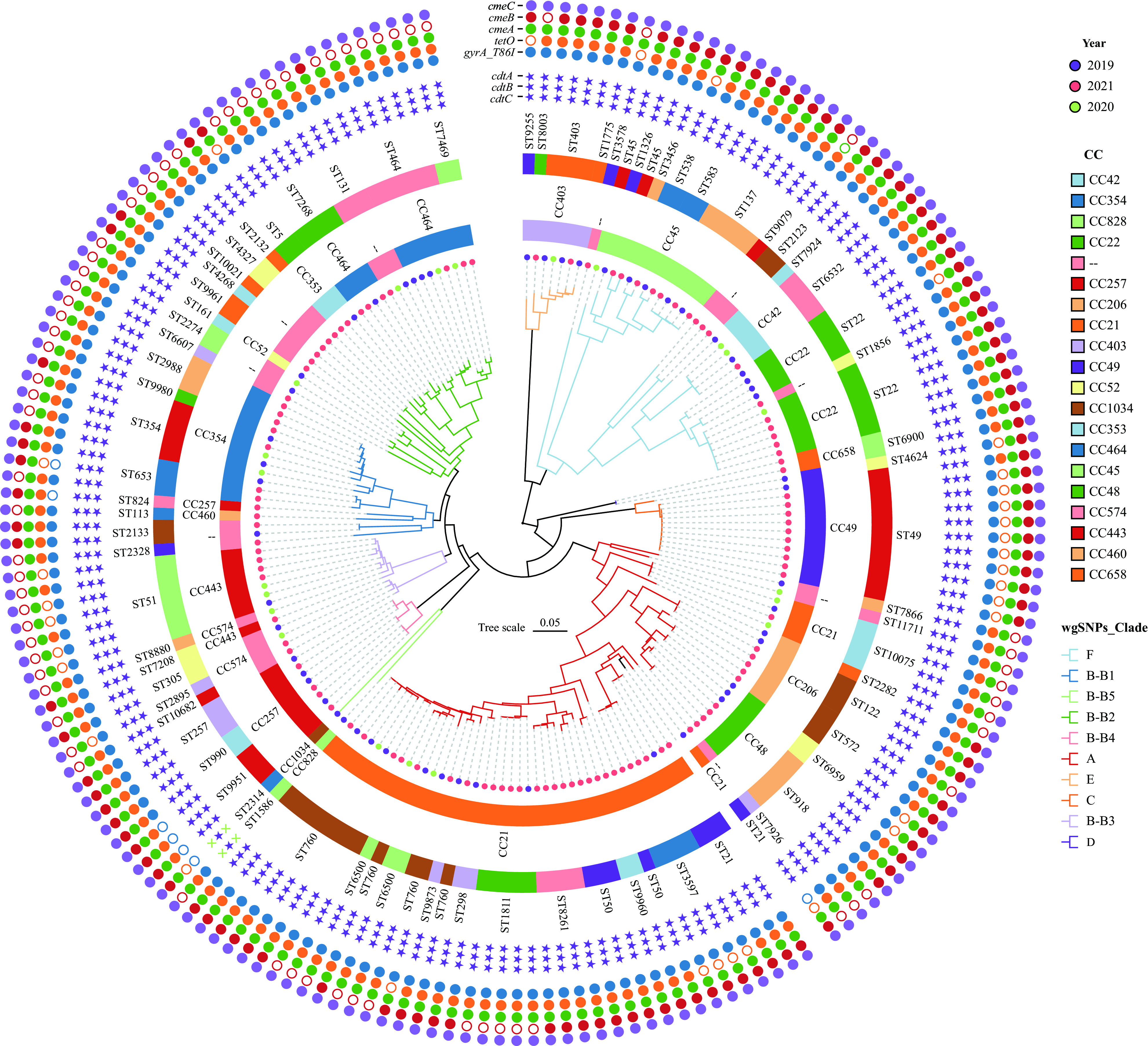
Results: Multilocus sequence typing analysis revealed high genetic diversity among the 184 C. jejuni strains, identifying 71 sequence types (STs) and 19 clonal complexes (CCs). The most prevalent ST was ST760 (6.5%), and the most common CC was CC21 (24.5%), consisting of 11 STs. High resistance rates were observed for ciprofloxacin (76.6%), nalidixic acid (76.1%), and tetracycline (71.2%). A total of 77 C. jejuni isolates (41.8%) exhibited multidrug resistance with 43 resistance patterns. Virulome analysis disclosed the differential distribution of virulence factors related to adherence, colonization, chemotaxis, as well as lipo-oligosaccharide and capsular polysaccharide biosynthesis. Resistome analysis demonstrated widespread resistance to quinolones and tetracycline, but low rates of macrolides resistance. The phylogeny, based on whole genome single nucleotide polymorphisms, indicated a high degree of clonality and grouped the C. jejuni strains into six clades. Closely related isolates that were part of a genetic cluster mostly shared a homogenous clonal complex.
Conclusions: The present study emphasizes the rising resistance to quinolones and tetracycline, as well as the virulence potential and diverse genotypes identified among C. jejuni strains isolated from diarrheal patients in Beijing.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


