{"title":"Effects of luseogliflozin treatment on hyperglycemia-induced muscle atrophy in rats.","authors":"Keyu Xie, Ken Sugimoto, Minoru Tanaka, Hiroshi Akasaka, Taku Fujimoto, Toshimasa Takahashi, Yuri Onishi, Tomohiro Minami, Shino Yoshida, Yoichi Takami, Koichi Yamamoto, Hiromi Rakugi","doi":"10.3164/jcbn.22-58","DOIUrl":null,"url":null,"abstract":"<p><p>Diabetes mellitus is recognized as a risk factor for sarcopenia. Luseogliflozin, a selective sodium-glucose cotransporter 2 (SGLT2) inhibitor, reduces inflammation and oxidative stress by improving hyperglycemia, subsequently improving hepatosteatosis or kidney dysfunction. However, the effects of SGLT2 inhibitor on the regulation of skeletal muscle mass or function in hyperglycemia are still unknown. In this study, we investigated the effects of luseogliflozin-mediated attenuation of hyperglycemia on the prevention of muscle atrophy. Twenty-four male Sprague-Dawley rats were randomly divided into four groups: control, control with SGLT2 inhibitor treatment, hyperglycemia, and hyperglycemia with SGLT2 inhibitor treatment. The hyperglycemic rodent model was established using a single injection of streptozotocin, a compound with preferential toxicity toward pancreatic beta cells. Muscle atrophy in streptozotocin-induced hyperglycemic model rats was inhibited by the suppression of hyperglycemia using luseogliflozin, which consequently suppressed hyperglycemia-mediated increase in the levels of advanced glycation end products (AGEs) and activated the protein degradation pathway in muscle cells. Treatment with luseogliflozin can restore the hyperglycemia-induced loss in the muscle mass to some degree partly through the inhibition of AGEs-induced or homeostatic disruption of mitochondria-induced activation of muscle degradation.</p>","PeriodicalId":15429,"journal":{"name":"Journal of Clinical Biochemistry and Nutrition","volume":"72 3","pages":"248-255"},"PeriodicalIF":2.0000,"publicationDate":"2023-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/6d/ac/jcbn22-58.PMC10209601.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Biochemistry and Nutrition","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3164/jcbn.22-58","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"NUTRITION & DIETETICS","Score":null,"Total":0}
引用次数: 0
Abstract
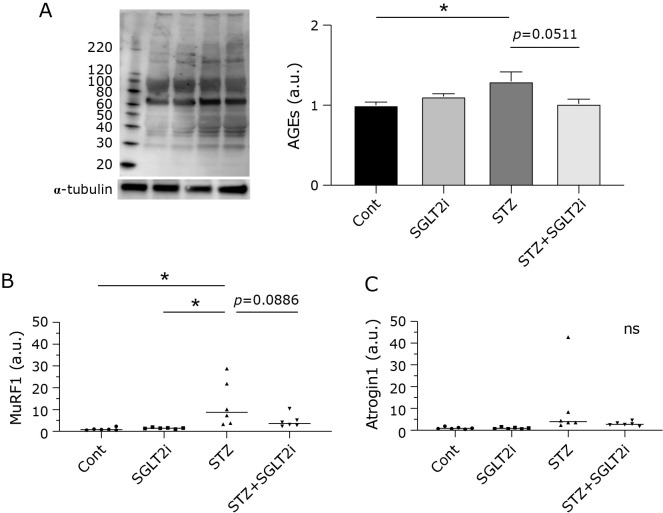
Diabetes mellitus is recognized as a risk factor for sarcopenia. Luseogliflozin, a selective sodium-glucose cotransporter 2 (SGLT2) inhibitor, reduces inflammation and oxidative stress by improving hyperglycemia, subsequently improving hepatosteatosis or kidney dysfunction. However, the effects of SGLT2 inhibitor on the regulation of skeletal muscle mass or function in hyperglycemia are still unknown. In this study, we investigated the effects of luseogliflozin-mediated attenuation of hyperglycemia on the prevention of muscle atrophy. Twenty-four male Sprague-Dawley rats were randomly divided into four groups: control, control with SGLT2 inhibitor treatment, hyperglycemia, and hyperglycemia with SGLT2 inhibitor treatment. The hyperglycemic rodent model was established using a single injection of streptozotocin, a compound with preferential toxicity toward pancreatic beta cells. Muscle atrophy in streptozotocin-induced hyperglycemic model rats was inhibited by the suppression of hyperglycemia using luseogliflozin, which consequently suppressed hyperglycemia-mediated increase in the levels of advanced glycation end products (AGEs) and activated the protein degradation pathway in muscle cells. Treatment with luseogliflozin can restore the hyperglycemia-induced loss in the muscle mass to some degree partly through the inhibition of AGEs-induced or homeostatic disruption of mitochondria-induced activation of muscle degradation.
期刊介绍:
Journal of Clinical Biochemistry and Nutrition (JCBN) is
an international, interdisciplinary publication encompassing
chemical, biochemical, physiological, pathological, toxicological and medical approaches to research on lipid peroxidation, free radicals, oxidative stress and nutrition. The
Journal welcomes original contributions dealing with all
aspects of clinical biochemistry and clinical nutrition
including both in vitro and in vivo studies.



 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


