{"title":"De novo myelodysplastic syndrome in a Rothmund-Thomson Syndrome patient with novel pathogenic <i>RECQL4</i> variants.","authors":"Chuanhe Jiang, Hao Zhang, Chuxian Zhao, Luxiang Wang, Xiaoxia Hu, Zengkai Pan","doi":"10.1097/BS9.0000000000000152","DOIUrl":null,"url":null,"abstract":"<p><p>Rothmund-Thomson syndrome (RTS) is a rare autosomal-recessive disorder with clinical features consisting of rash, poikiloderma, sparse hair, short stature, juvenile cataracts, skeletal abnormalities, and cancer predisposition. Genetic studies involving detection of pathogenic <i>RECQL4</i> variants provide the diagnostic certitude. Osteosarcoma was found in two-thirds <i>RECQL4</i>-mutated RTS patients, while hematological malignancies were rarely reported. The variant diversity of <i>RECQL4</i> gene has not been fully identified and mutations associated with hematologic malignancies are not well described. In this study, we presented a pedigree of RTS from a Chinese family, among which the proband was diagnosed with de novo myelodysplastic syndrome (MDS). Comprehensive medical examination and chromosome karyotyping were performed on the proband. Whole exome sequencing (WES) was performed on the proband, his sister and his mother. The familial cosegregation of sequence variants derived from WES was conducted by polymerase chain reaction-based Sanger sequencing. Structures of candidate RECQL4 mutants were done by in silico analysis to assess pathogenicity. Three novel <i>RECQL4</i> germline variants, including c.T274C, c.G3014A, and c.G801C, were identified by WES and validated by Sanger sequencing. Prediction of conformation indicated that the structural stability of human RECQL4 protein was largely affected with these variants. The co-occurring <i>U2AF1</i> p.S34F and <i>TP53</i> p.Y220C mutations might contribute to the development of MDS. Our study expands the mutational spectrum of <i>RECQL4</i> and provides underlying molecular mechanism for the development of MDS in RTS patients.</p>","PeriodicalId":67343,"journal":{"name":"血液科学(英文)","volume":"5 2","pages":"125-130"},"PeriodicalIF":2.7000,"publicationDate":"2023-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/0a/7a/bs9-5-125.PMC10205365.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"血液科学(英文)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1097/BS9.0000000000000152","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
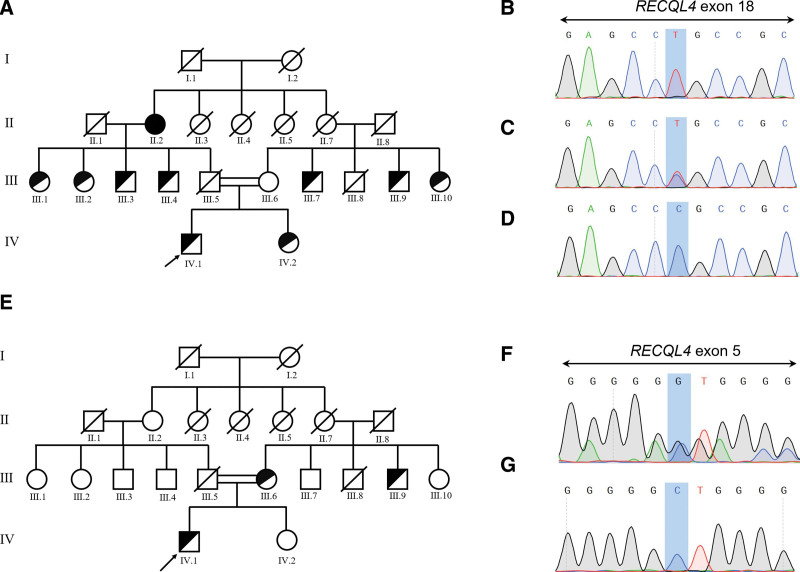
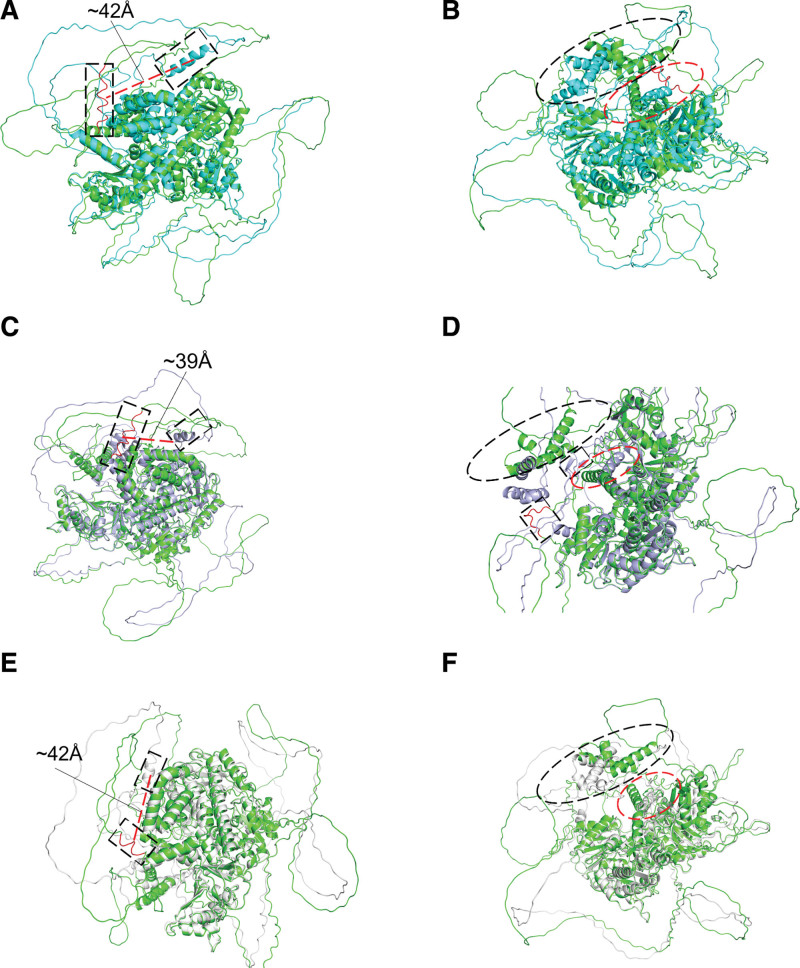
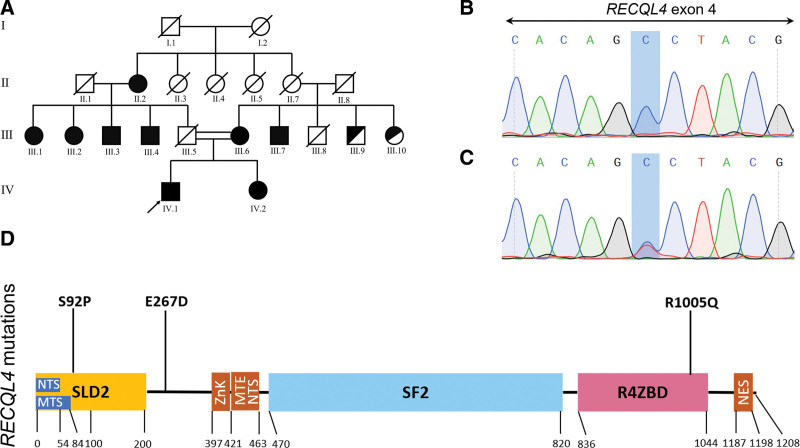
Rothmund-Thomson syndrome (RTS) is a rare autosomal-recessive disorder with clinical features consisting of rash, poikiloderma, sparse hair, short stature, juvenile cataracts, skeletal abnormalities, and cancer predisposition. Genetic studies involving detection of pathogenic RECQL4 variants provide the diagnostic certitude. Osteosarcoma was found in two-thirds RECQL4-mutated RTS patients, while hematological malignancies were rarely reported. The variant diversity of RECQL4 gene has not been fully identified and mutations associated with hematologic malignancies are not well described. In this study, we presented a pedigree of RTS from a Chinese family, among which the proband was diagnosed with de novo myelodysplastic syndrome (MDS). Comprehensive medical examination and chromosome karyotyping were performed on the proband. Whole exome sequencing (WES) was performed on the proband, his sister and his mother. The familial cosegregation of sequence variants derived from WES was conducted by polymerase chain reaction-based Sanger sequencing. Structures of candidate RECQL4 mutants were done by in silico analysis to assess pathogenicity. Three novel RECQL4 germline variants, including c.T274C, c.G3014A, and c.G801C, were identified by WES and validated by Sanger sequencing. Prediction of conformation indicated that the structural stability of human RECQL4 protein was largely affected with these variants. The co-occurring U2AF1 p.S34F and TP53 p.Y220C mutations might contribute to the development of MDS. Our study expands the mutational spectrum of RECQL4 and provides underlying molecular mechanism for the development of MDS in RTS patients.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


