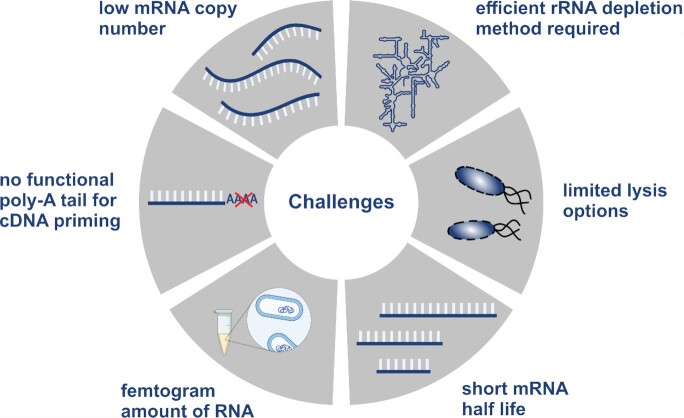
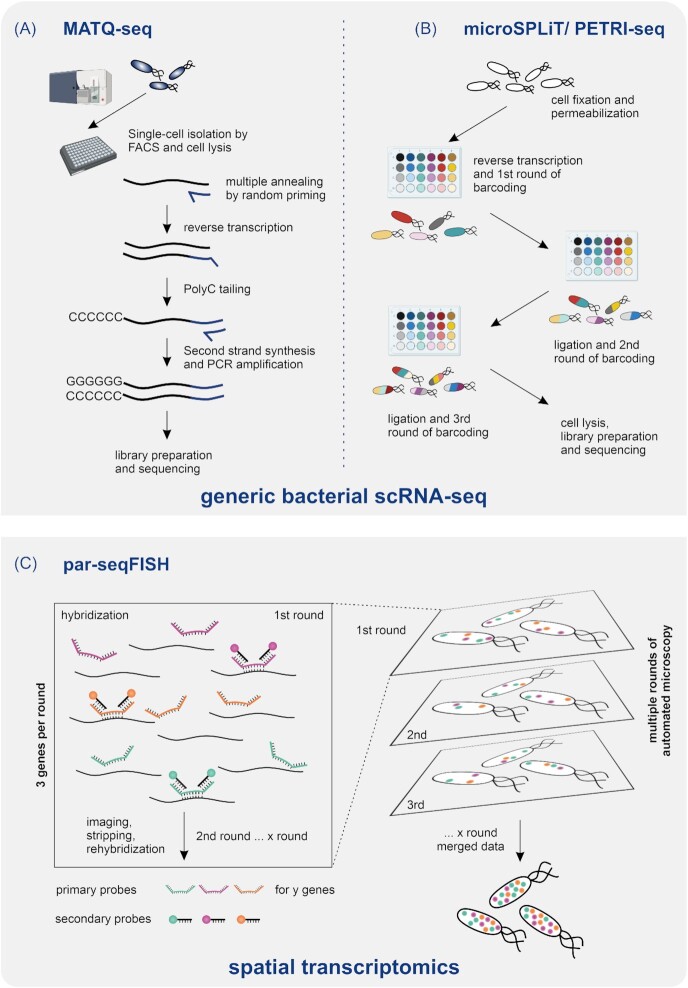
{"title":"Ushering in a new era of single-cell transcriptomics in bacteria.","authors":"Christina Homberger, Lars Barquist, Jörg Vogel","doi":"10.1093/femsml/uqac020","DOIUrl":null,"url":null,"abstract":"<p><p>Transcriptome analysis of individual cells by single-cell RNA-seq (scRNA-seq) has become routine for eukaryotic tissues, even being applied to whole multicellular organisms. In contrast, developing methods to read the transcriptome of single bacterial cells has proven more challenging, despite a general perception of bacteria as much simpler than eukaryotes. Bacterial cells are harder to lyse, their RNA content is about two orders of magnitude lower than that of eukaryotic cells, and bacterial mRNAs are less stable than their eukaryotic counterparts. Most importantly, bacterial transcripts lack functional poly(A) tails, precluding simple adaptation of popular standard eukaryotic scRNA-seq protocols that come with the double advantage of specific mRNA amplification and concomitant depletion of rRNA. However, thanks to very recent breakthroughs in methodology, bacterial scRNA-seq is now feasible. This short review will discuss recently published bacterial scRNA-seq approaches (MATQ-seq, microSPLiT, and PETRI-seq) and a spatial transcriptomics approach based on multiplexed <i>in situ</i> hybridization (par-seqFISH). Together, these novel approaches will not only enable a new understanding of cell-to-cell variation in bacterial gene expression, they also promise a new microbiology by enabling high-resolution profiling of gene activity in complex microbial consortia such as the microbiome or pathogens as they invade, replicate, and persist in host tissue.</p>","PeriodicalId":74189,"journal":{"name":"microLife","volume":"3 ","pages":"uqac020"},"PeriodicalIF":0.0000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10117829/pdf/","citationCount":"13","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"microLife","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/femsml/uqac020","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 13
Abstract
Transcriptome analysis of individual cells by single-cell RNA-seq (scRNA-seq) has become routine for eukaryotic tissues, even being applied to whole multicellular organisms. In contrast, developing methods to read the transcriptome of single bacterial cells has proven more challenging, despite a general perception of bacteria as much simpler than eukaryotes. Bacterial cells are harder to lyse, their RNA content is about two orders of magnitude lower than that of eukaryotic cells, and bacterial mRNAs are less stable than their eukaryotic counterparts. Most importantly, bacterial transcripts lack functional poly(A) tails, precluding simple adaptation of popular standard eukaryotic scRNA-seq protocols that come with the double advantage of specific mRNA amplification and concomitant depletion of rRNA. However, thanks to very recent breakthroughs in methodology, bacterial scRNA-seq is now feasible. This short review will discuss recently published bacterial scRNA-seq approaches (MATQ-seq, microSPLiT, and PETRI-seq) and a spatial transcriptomics approach based on multiplexed in situ hybridization (par-seqFISH). Together, these novel approaches will not only enable a new understanding of cell-to-cell variation in bacterial gene expression, they also promise a new microbiology by enabling high-resolution profiling of gene activity in complex microbial consortia such as the microbiome or pathogens as they invade, replicate, and persist in host tissue.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


