SMPD1 expression profile and mutation landscape help decipher genotype-phenotype association and precision diagnosis for acid sphingomyelinase deficiency.
Ruisong Wang, Ziyi Qin, Long Huang, Huiling Luo, Han Peng, Xinyu Zhou, Zhixiang Zhao, Mingyao Liu, Pinhong Yang, Tieliu Shi
{"title":"SMPD1 expression profile and mutation landscape help decipher genotype-phenotype association and precision diagnosis for acid sphingomyelinase deficiency.","authors":"Ruisong Wang, Ziyi Qin, Long Huang, Huiling Luo, Han Peng, Xinyu Zhou, Zhixiang Zhao, Mingyao Liu, Pinhong Yang, Tieliu Shi","doi":"10.1186/s41065-023-00272-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Acid sphingomyelinase deficiency (ASMD) disorder, also known as Niemann-Pick disease (NPD) is a rare genetic disease caused by mutations in SMPD1 gene, which encodes sphingomyelin phosphodiesterase (ASM). Except for liver and spleen enlargement and lung disease, two subtypes (Type A and B) of NDP have different onset times, survival times, ASM activities, and neurological abnormalities. To comprehensively explore NPD's genotype-phenotype association and pathophysiological characteristics, we collected 144 NPD cases with strict quality control through literature mining.</p><p><strong>Results: </strong>The difference in ASM activity can differentiate NPD type A from other subtypes, with the ratio of ASM activity to the reference values being lower in type A (threshold 0.045 (4.45%)). Severe variations, such as deletion and insertion, can cause complete loss of ASM function, leading to type A, whereas relatively mild missense mutations generally result in type B. Among reported mutations, the p.Arg3AlafsX76 mutation is highly prevalent in the Chinese population, and the p.R608del mutation is common in Mediterranean countries. The expression profiles of SMPD1 from GTEx and single-cell RNA sequencing data of multiple fetal tissues showed that high expressions of SMPD1 can be observed in the liver, spleen, and brain tissues of adults and hepatoblasts, hematopoietic stem cells, STC2_TLX1-positive cells, mesothelial cells of the spleen, vascular endothelial cells of the cerebellum and the cerebrum of fetuses, indicating that SMPD1 dysfunction is highly likely to have a significant effect on the function of those cell types during development and the clinicians need pay attention to these organs or tissues as well during diagnosis. In addition, we also predicted 21 new pathogenic mutations in the SMPD1 gene that potentially cause the NPD, signifying that more rare cases will be detected with those mutations in SMPD1. Finally, we also analysed the function of the NPD type A cells following the extracellular milieu.</p><p><strong>Conclusions: </strong>Our study is the first to elucidate the effects of SMPD1 mutation on cell types and at the tissue level, which provides new insights into the genotype-phenotype association and can help in the precise diagnosis of NPD.</p>","PeriodicalId":12862,"journal":{"name":"Hereditas","volume":"160 1","pages":"11"},"PeriodicalIF":2.7000,"publicationDate":"2023-03-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10009935/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hereditas","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s41065-023-00272-1","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Acid sphingomyelinase deficiency (ASMD) disorder, also known as Niemann-Pick disease (NPD) is a rare genetic disease caused by mutations in SMPD1 gene, which encodes sphingomyelin phosphodiesterase (ASM). Except for liver and spleen enlargement and lung disease, two subtypes (Type A and B) of NDP have different onset times, survival times, ASM activities, and neurological abnormalities. To comprehensively explore NPD's genotype-phenotype association and pathophysiological characteristics, we collected 144 NPD cases with strict quality control through literature mining.
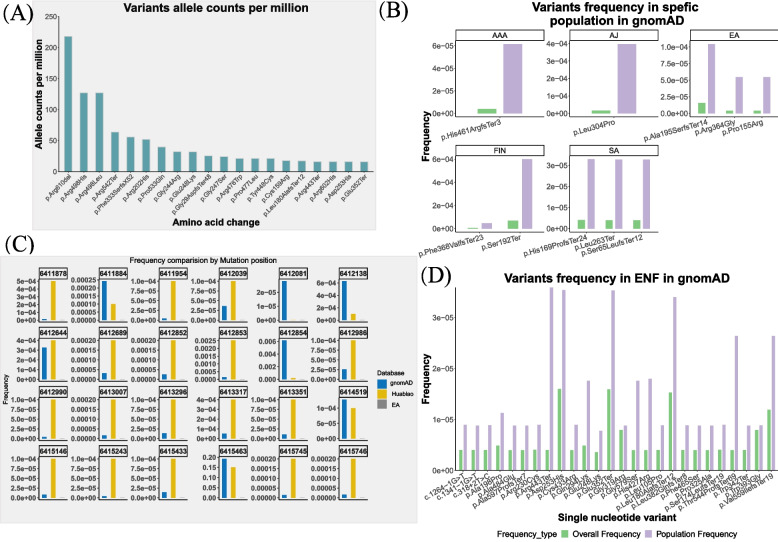
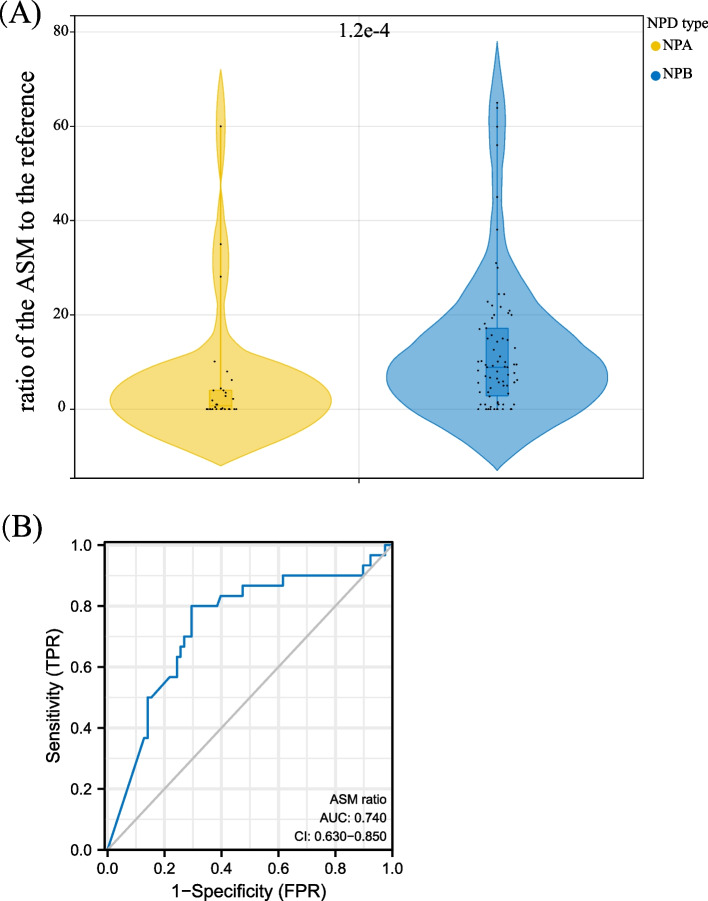
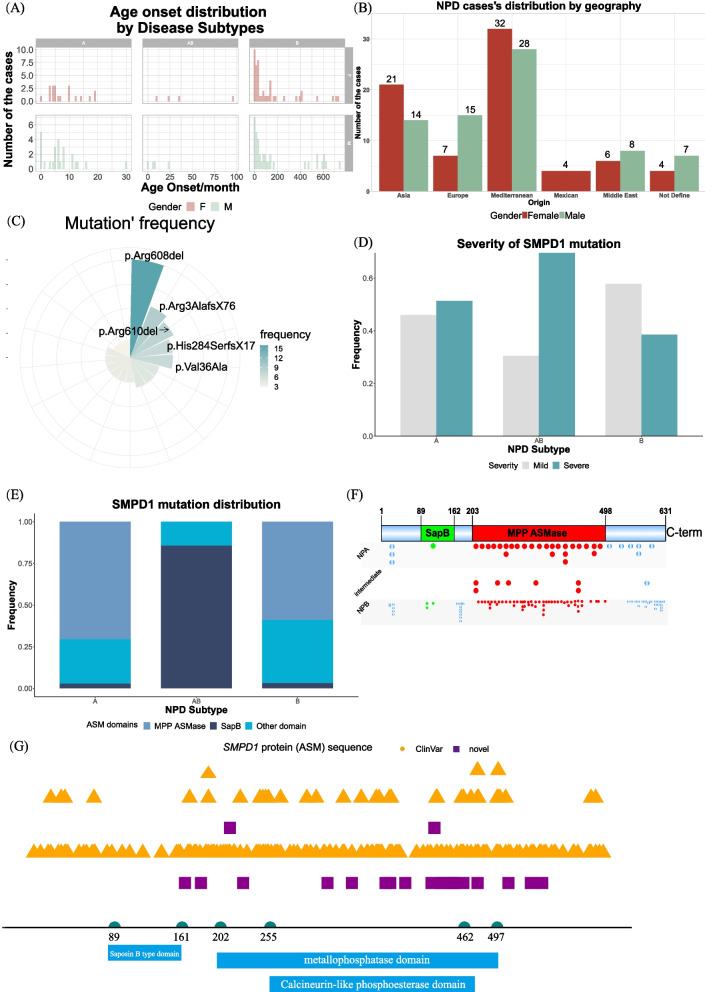
Results: The difference in ASM activity can differentiate NPD type A from other subtypes, with the ratio of ASM activity to the reference values being lower in type A (threshold 0.045 (4.45%)). Severe variations, such as deletion and insertion, can cause complete loss of ASM function, leading to type A, whereas relatively mild missense mutations generally result in type B. Among reported mutations, the p.Arg3AlafsX76 mutation is highly prevalent in the Chinese population, and the p.R608del mutation is common in Mediterranean countries. The expression profiles of SMPD1 from GTEx and single-cell RNA sequencing data of multiple fetal tissues showed that high expressions of SMPD1 can be observed in the liver, spleen, and brain tissues of adults and hepatoblasts, hematopoietic stem cells, STC2_TLX1-positive cells, mesothelial cells of the spleen, vascular endothelial cells of the cerebellum and the cerebrum of fetuses, indicating that SMPD1 dysfunction is highly likely to have a significant effect on the function of those cell types during development and the clinicians need pay attention to these organs or tissues as well during diagnosis. In addition, we also predicted 21 new pathogenic mutations in the SMPD1 gene that potentially cause the NPD, signifying that more rare cases will be detected with those mutations in SMPD1. Finally, we also analysed the function of the NPD type A cells following the extracellular milieu.
Conclusions: Our study is the first to elucidate the effects of SMPD1 mutation on cell types and at the tissue level, which provides new insights into the genotype-phenotype association and can help in the precise diagnosis of NPD.
HereditasBiochemistry, Genetics and Molecular Biology-Genetics
CiteScore
3.80
自引率
3.70%
发文量
0
期刊介绍:
For almost a century, Hereditas has published original cutting-edge research and reviews. As the Official journal of the Mendelian Society of Lund, the journal welcomes research from across all areas of genetics and genomics. Topics of interest include human and medical genetics, animal and plant genetics, microbial genetics, agriculture and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


