Megane Delourme, Chaix Charlene, Laurene Gerard, Benjamin Ganne, Pierre Perrin, Catherine Vovan, Karine Bertaux, Karine Nguyen, Rafaëlle Bernard, Frederique Magdinier
{"title":"Complex 4q35 and 10q26 Rearrangements: A Challenge for Molecular Diagnosis of Patients With Facioscapulohumeral Dystrophy.","authors":"Megane Delourme, Chaix Charlene, Laurene Gerard, Benjamin Ganne, Pierre Perrin, Catherine Vovan, Karine Bertaux, Karine Nguyen, Rafaëlle Bernard, Frederique Magdinier","doi":"10.1212/NXG.0000000000200076","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong>After clinical evaluation, the molecular diagnosis of type 1 facioscapulohumeral dystrophy (FSHD1) relies in most laboratories on the detection of a shortened D4Z4 array at the 4q35 locus by Southern blotting. In many instances, this molecular diagnosis remains inconclusive and requires additional experiments to determine the number of D4Z4 units or identify somatic mosaicism, 4q-10q translocations, and proximal p13E-11 deletions. These limitations highlight the need for alternative methodologies, illustrated by the recent emergence of novel technologies such as molecular combing (MC), single molecule optical mapping (SMOM), or Oxford Nanopore-based long-read sequencing providing a more comprehensive analysis of 4q and 10q loci. Over the last decade, MC revealed a further increasing complexity in the organization of the 4q and 10q distal regions in patients with FSHD with <i>cis</i>-duplication of D4Z4 arrays in approximately 1%-2% of cases.</p><p><strong>Methods: </strong>By using MC, we investigated in our center 2,363 cases for molecular diagnosis of FSHD. We also evaluated whether previously reported <i>cis</i>-duplications might be identified by SMOM using the Bionano EnFocus FSHD 1.0 algorithm.</p><p><strong>Results: </strong>In our cohort of 2,363 samples, we identified 147 individuals carrying an atypical organization of the 4q35 or 10q26 loci. Mosaicism is the most frequent category followed by <i>cis</i>-duplications of the D4Z4 array. We report here chromosomal abnormalities of the 4q35 or 10q26 loci in 54 patients clinically described as FSHD, which are not present in the healthy population. In one-third of the 54 patients, these rearrangements are the only genetic defect suggesting that they might be causative of the disease. By analyzing DNA samples from 3 patients carrying a complex rearrangement of the 4q35 region, we further showed that the SMOM direct assembly of the 4q and 10q alleles failed to reveal these abnormalities and lead to negative results for FSHD molecular diagnosis.</p><p><strong>Discussion: </strong>This work further highlights the complexity of the 4q and 10q subtelomeric regions and the need of in-depth analyses in a significant number of cases. This work also highlights the complexity of the 4q35 region and interpretation issues with consequences on the molecular diagnosis of patients or genetic counseling.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"9 3","pages":"e200076"},"PeriodicalIF":3.0000,"publicationDate":"2023-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10188231/pdf/","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200076","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 2
Abstract
Background and objectives: After clinical evaluation, the molecular diagnosis of type 1 facioscapulohumeral dystrophy (FSHD1) relies in most laboratories on the detection of a shortened D4Z4 array at the 4q35 locus by Southern blotting. In many instances, this molecular diagnosis remains inconclusive and requires additional experiments to determine the number of D4Z4 units or identify somatic mosaicism, 4q-10q translocations, and proximal p13E-11 deletions. These limitations highlight the need for alternative methodologies, illustrated by the recent emergence of novel technologies such as molecular combing (MC), single molecule optical mapping (SMOM), or Oxford Nanopore-based long-read sequencing providing a more comprehensive analysis of 4q and 10q loci. Over the last decade, MC revealed a further increasing complexity in the organization of the 4q and 10q distal regions in patients with FSHD with cis-duplication of D4Z4 arrays in approximately 1%-2% of cases.
Methods: By using MC, we investigated in our center 2,363 cases for molecular diagnosis of FSHD. We also evaluated whether previously reported cis-duplications might be identified by SMOM using the Bionano EnFocus FSHD 1.0 algorithm.
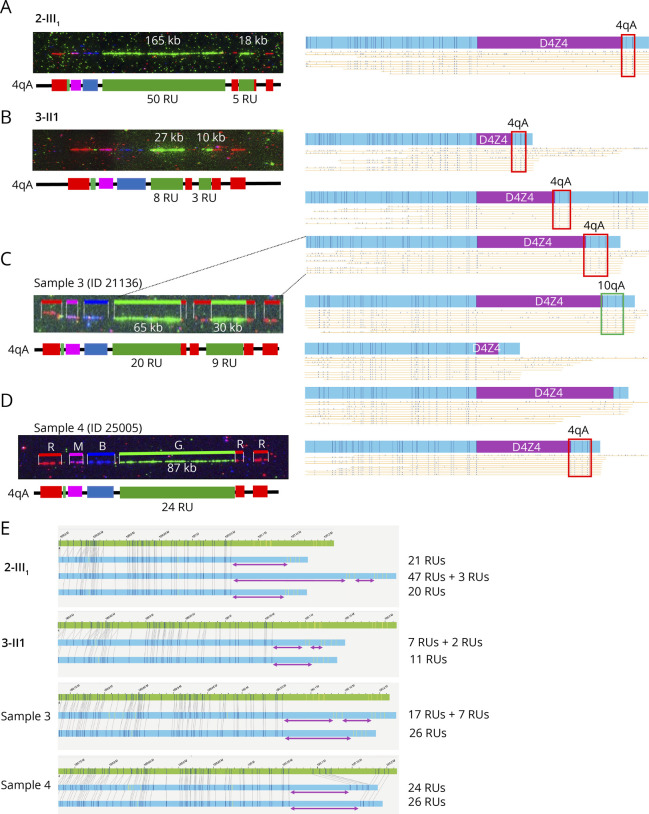
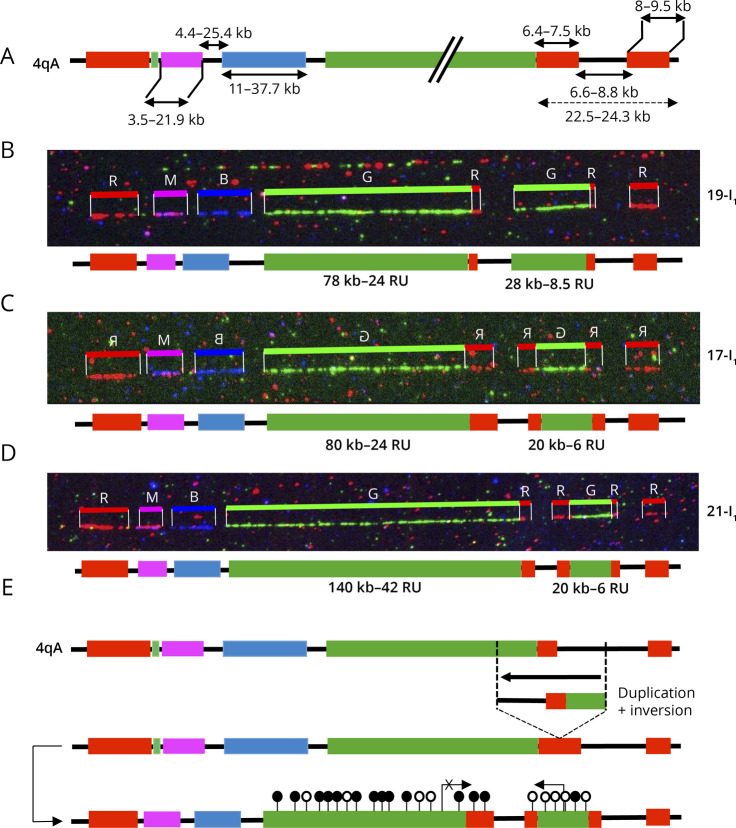
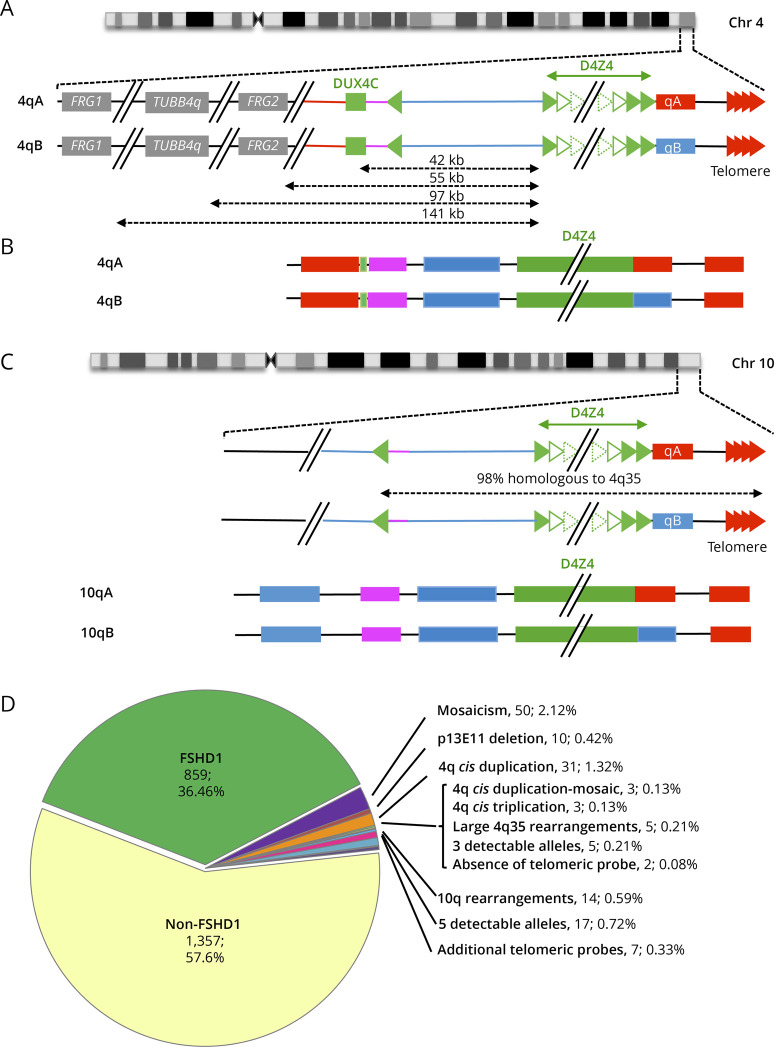
Results: In our cohort of 2,363 samples, we identified 147 individuals carrying an atypical organization of the 4q35 or 10q26 loci. Mosaicism is the most frequent category followed by cis-duplications of the D4Z4 array. We report here chromosomal abnormalities of the 4q35 or 10q26 loci in 54 patients clinically described as FSHD, which are not present in the healthy population. In one-third of the 54 patients, these rearrangements are the only genetic defect suggesting that they might be causative of the disease. By analyzing DNA samples from 3 patients carrying a complex rearrangement of the 4q35 region, we further showed that the SMOM direct assembly of the 4q and 10q alleles failed to reveal these abnormalities and lead to negative results for FSHD molecular diagnosis.
Discussion: This work further highlights the complexity of the 4q and 10q subtelomeric regions and the need of in-depth analyses in a significant number of cases. This work also highlights the complexity of the 4q35 region and interpretation issues with consequences on the molecular diagnosis of patients or genetic counseling.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


