A Computational Study on Selected Alkaloids as SARS-CoV-2 Inhibitors: PASS Prediction, Molecular Docking, ADMET Analysis, DFT, and Molecular Dynamics Simulations.
Md Golam Mortuza, Md Abul Hasan Roni, Ajoy Kumer, Suvro Biswas, Md Abu Saleh, Shirmin Islam, Samia Sadaf, Fahmida Akther
{"title":"A Computational Study on Selected Alkaloids as SARS-CoV-2 Inhibitors: PASS Prediction, Molecular Docking, ADMET Analysis, DFT, and Molecular Dynamics Simulations.","authors":"Md Golam Mortuza, Md Abul Hasan Roni, Ajoy Kumer, Suvro Biswas, Md Abu Saleh, Shirmin Islam, Samia Sadaf, Fahmida Akther","doi":"10.1155/2023/9975275","DOIUrl":null,"url":null,"abstract":"<p><p>Despite treatments and vaccinations, it remains difficult to develop naturally occurring COVID-19 inhibitors. Here, our main objective is to find potential lead compounds from the retrieved alkaloids with antiviral and other biological properties that selectively target the main SARS-CoV-2 protease (<i>M</i><sup>pro</sup>), which is required for viral replication. In this work, 252 alkaloids were aligned using Lipinski's rule of five and their antiviral activity was then assessed. The prediction of activity spectrum of substances (PASS) data was used to confirm the antiviral activities of 112 alkaloids. Finally, 50 alkaloids were docked with <i>M</i><sup>pro</sup>. Furthermore, assessments of molecular electrostatic potential surface (MEPS), density functional theory (DFT), and absorption, distribution, metabolism, excretion, and toxicity (ADMET) were performed, and a few of them appeared to have potential as candidates for oral administration. Molecular dynamics simulations (MDS) with a time step of up to 100 ns were used to confirm that the three docked complexes were more stable. It was found that the most prevalent and active binding sites that limit <i>M</i><sup>pro</sup>'sactivity are PHE294, ARG298, and GLN110. All retrieved data were compared to conventional antivirals, fumarostelline, strychnidin-10-one (L-1), 2,3-dimethoxy-brucin (L-7), and alkaloid ND-305B (L-16) and were proposed as enhanced SARS-CoV-2 inhibitors. Finally, with additional clinical or necessary study, it may be able to use these indicated natural alkaloids or their analogs as potential therapeutic candidates.</p>","PeriodicalId":8826,"journal":{"name":"Biochemistry Research International","volume":"2023 ","pages":"9975275"},"PeriodicalIF":3.4000,"publicationDate":"2023-05-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10171978/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biochemistry Research International","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2023/9975275","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
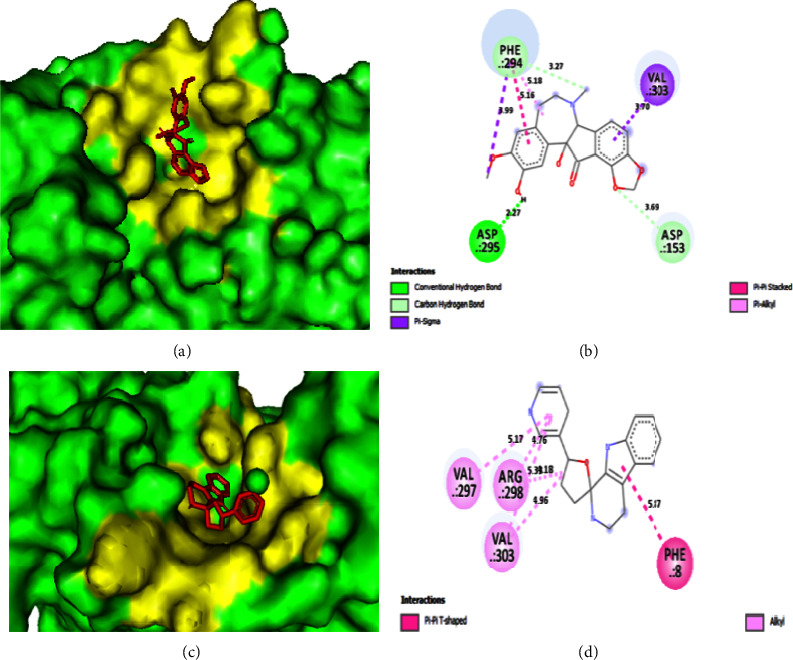
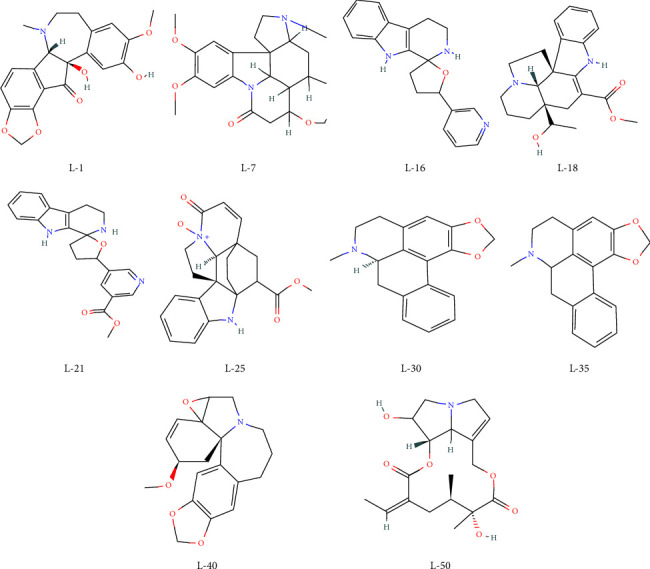
Despite treatments and vaccinations, it remains difficult to develop naturally occurring COVID-19 inhibitors. Here, our main objective is to find potential lead compounds from the retrieved alkaloids with antiviral and other biological properties that selectively target the main SARS-CoV-2 protease (Mpro), which is required for viral replication. In this work, 252 alkaloids were aligned using Lipinski's rule of five and their antiviral activity was then assessed. The prediction of activity spectrum of substances (PASS) data was used to confirm the antiviral activities of 112 alkaloids. Finally, 50 alkaloids were docked with Mpro. Furthermore, assessments of molecular electrostatic potential surface (MEPS), density functional theory (DFT), and absorption, distribution, metabolism, excretion, and toxicity (ADMET) were performed, and a few of them appeared to have potential as candidates for oral administration. Molecular dynamics simulations (MDS) with a time step of up to 100 ns were used to confirm that the three docked complexes were more stable. It was found that the most prevalent and active binding sites that limit Mpro'sactivity are PHE294, ARG298, and GLN110. All retrieved data were compared to conventional antivirals, fumarostelline, strychnidin-10-one (L-1), 2,3-dimethoxy-brucin (L-7), and alkaloid ND-305B (L-16) and were proposed as enhanced SARS-CoV-2 inhibitors. Finally, with additional clinical or necessary study, it may be able to use these indicated natural alkaloids or their analogs as potential therapeutic candidates.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


