{"title":"Camurati-Engelmann Disease: A Case-Based Review About an Ultrarare Bone Dysplasia.","authors":"Philipp Klemm, Iris Aykara, Uwe Lange","doi":"10.5152/eurjrheum.2023.21115","DOIUrl":null,"url":null,"abstract":"<p><p>Camurati-Engelmann disease or progressive diaphyseal dysplasia is a rare hereditary disease that results in a symmetrical hyperostosis of the long bones (cortical thickening) and/or the base of the skull. Camurati-Engelmann disease is also associated with myopathy and neurological manifestations. Clinically, Camurati-Engelmann disease typically presents with bone pain in the lower extremities, muscle weakness, and a wobbly, stilted gait. The disease is caused by mutations in the transforming growth factor-beta 1 gene. Up to date, about 300 cases have been described in the literature. In this case-based review, we present the clinical picture and genetic and radiological findings in a 20-yearold male patient we diagnosed with Camurati-Engelmann disease and our considerations in his treatment and compare the case to the literature. The diagnosis of Camurati-Engelmann disease was confirmed on patients' history, clinical and radiological findings, and genetic testing for transforming growth factor beta-1 mutation. The patient responded well to single therapy with zoledronic acid. Early diagnosis leads to improved clinical outcomes and increased quality of life in affected patients.</p>","PeriodicalId":12066,"journal":{"name":"European journal of rheumatology","volume":"10 1","pages":"34-38"},"PeriodicalIF":1.8000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/93/e7/ejr-10-1-34.PMC10152113.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European journal of rheumatology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5152/eurjrheum.2023.21115","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"RHEUMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
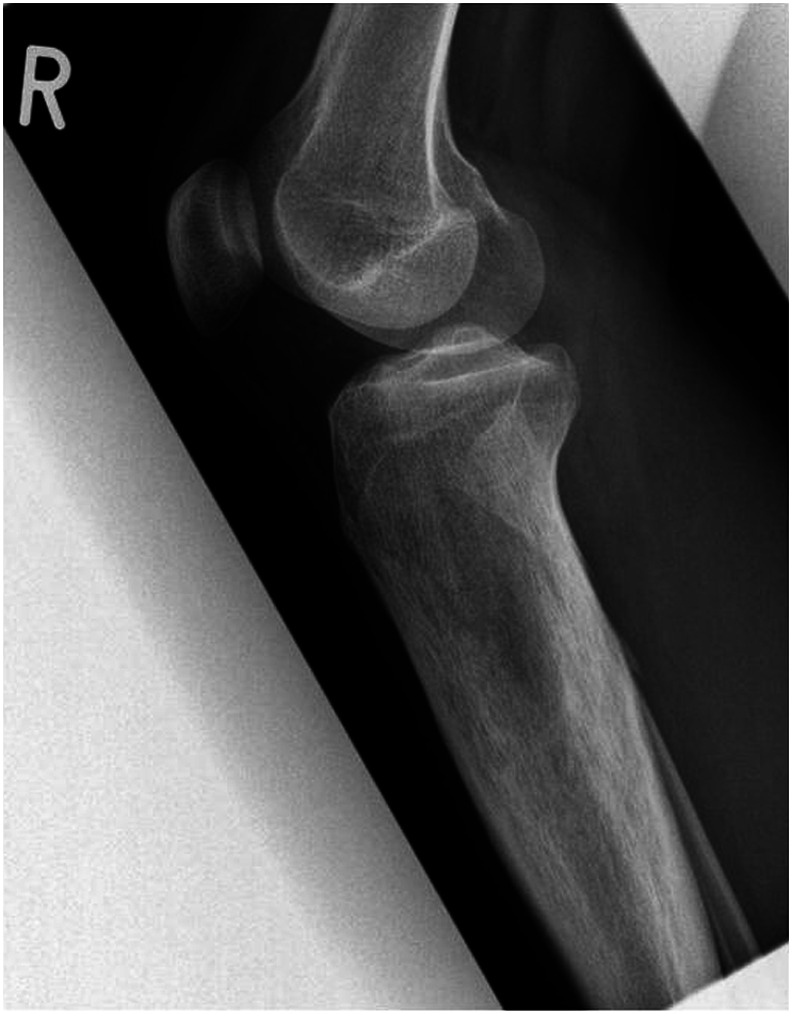
Camurati-Engelmann disease or progressive diaphyseal dysplasia is a rare hereditary disease that results in a symmetrical hyperostosis of the long bones (cortical thickening) and/or the base of the skull. Camurati-Engelmann disease is also associated with myopathy and neurological manifestations. Clinically, Camurati-Engelmann disease typically presents with bone pain in the lower extremities, muscle weakness, and a wobbly, stilted gait. The disease is caused by mutations in the transforming growth factor-beta 1 gene. Up to date, about 300 cases have been described in the literature. In this case-based review, we present the clinical picture and genetic and radiological findings in a 20-yearold male patient we diagnosed with Camurati-Engelmann disease and our considerations in his treatment and compare the case to the literature. The diagnosis of Camurati-Engelmann disease was confirmed on patients' history, clinical and radiological findings, and genetic testing for transforming growth factor beta-1 mutation. The patient responded well to single therapy with zoledronic acid. Early diagnosis leads to improved clinical outcomes and increased quality of life in affected patients.



 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


