Claudia Ciaccio, Chiara Pantaleoni, Marco Moscatelli, Luisa Chiapparini, Vincenzo Nigro, Enza Maria Valente, Francesca Sciacca, Laura Canafoglia, Sara Bulgheroni, Stefano D'Arrigo
{"title":"Neurologic, Neuropsychologic, and Neuroradiologic Features of <i>EBF3</i>-Related Syndrome.","authors":"Claudia Ciaccio, Chiara Pantaleoni, Marco Moscatelli, Luisa Chiapparini, Vincenzo Nigro, Enza Maria Valente, Francesca Sciacca, Laura Canafoglia, Sara Bulgheroni, Stefano D'Arrigo","doi":"10.1212/NXG.0000000000200049","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong>Heterozygous mutations or deletions of the <i>EBF3</i> gene are known to cause a syndrome characterized by intellectual disability, neurodevelopmental disorders, facial dysmorphisms, hypotonia, and ataxia; the latter is quite common despite in most patients brain MRI is reported to be normal. Despite the predominant neurologic involvement of <i>EBF3</i>-related syndrome, a systematic definition of neurologic, cognitive/behavioral, and neuroradiologic features is lacking.</p><p><strong>Methods: </strong>We report on 6 patients (2 females and 4 males, age range 2-12 years), of whom 4 carrying a heterozygous point mutation of the <i>EBF3</i> gene and 2 with 10q26 deletion encompassing the gene, diagnosed at Carlo Besta Neurologic Institute of Milan, Italy. Clinical evaluation was performed by a pediatric neurologist and pediatric dysmorphologist; ataxia severity was rated by Scale for the Assessment and Rating of Ataxia (SARA); brain MRIs were reviewed by expert neuroradiologists; general quotient levels were obtained through standardized Griffiths Mental Development Scales. Patients carrying a 10q26.3 deletion were diagnosed by array-CGH, whereas <i>EBF3</i> variants were detected by whole exome sequencing.</p><p><strong>Results: </strong>Phenotype was consistent in all patients, but with wide variability in severity. Developmental milestones were invariably delayed and resulted in an extremely variable cognitive impairment. All patients showed ataxic signs, as confirmed by SARA scores, often associated with hypotonia. Brain MRI revealed in all children a cerebellar malformation with vermis hypoplasia and a peculiar foliation anomaly characterized by a radial disposition of cerebellar folia (dandelion sign). Neurophysiologic examinations were unremarkable.</p><p><strong>Discussion: </strong><i>EBF3</i>-related syndrome has been so far described as a neurodevelopmental condition with dysmorphic traits, with limited emphasis on the neurologic features; we highlight the predominant neurologic involvement of these patients, which can be explained at least in part by the underlying cerebellar malformation. We therefore propose that <i>EBF3</i>-related syndrome should be classified and treated as a congenital, nonprogressive ataxia.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"9 2","pages":"e200049"},"PeriodicalIF":3.0000,"publicationDate":"2023-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/98/da/NXG-2022-200052.PMC10117703.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200049","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
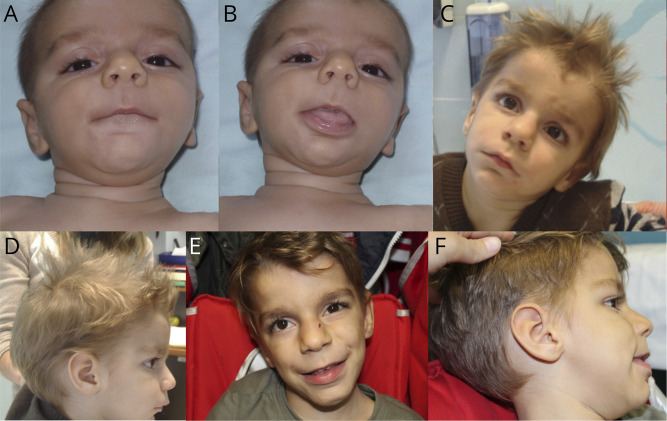
Background and objectives: Heterozygous mutations or deletions of the EBF3 gene are known to cause a syndrome characterized by intellectual disability, neurodevelopmental disorders, facial dysmorphisms, hypotonia, and ataxia; the latter is quite common despite in most patients brain MRI is reported to be normal. Despite the predominant neurologic involvement of EBF3-related syndrome, a systematic definition of neurologic, cognitive/behavioral, and neuroradiologic features is lacking.
Methods: We report on 6 patients (2 females and 4 males, age range 2-12 years), of whom 4 carrying a heterozygous point mutation of the EBF3 gene and 2 with 10q26 deletion encompassing the gene, diagnosed at Carlo Besta Neurologic Institute of Milan, Italy. Clinical evaluation was performed by a pediatric neurologist and pediatric dysmorphologist; ataxia severity was rated by Scale for the Assessment and Rating of Ataxia (SARA); brain MRIs were reviewed by expert neuroradiologists; general quotient levels were obtained through standardized Griffiths Mental Development Scales. Patients carrying a 10q26.3 deletion were diagnosed by array-CGH, whereas EBF3 variants were detected by whole exome sequencing.
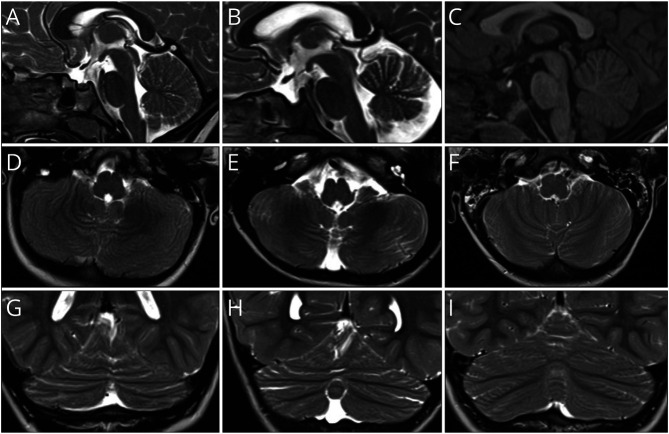
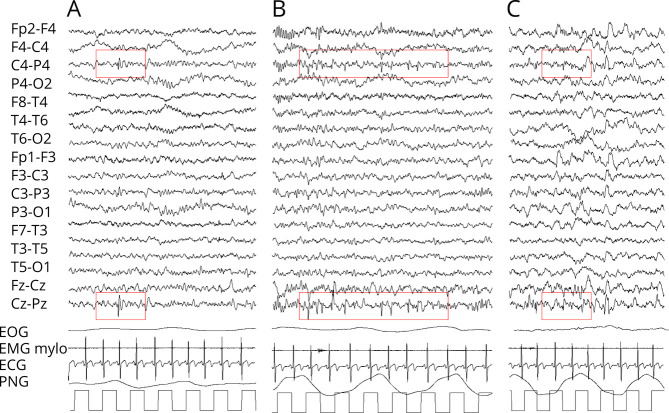
Results: Phenotype was consistent in all patients, but with wide variability in severity. Developmental milestones were invariably delayed and resulted in an extremely variable cognitive impairment. All patients showed ataxic signs, as confirmed by SARA scores, often associated with hypotonia. Brain MRI revealed in all children a cerebellar malformation with vermis hypoplasia and a peculiar foliation anomaly characterized by a radial disposition of cerebellar folia (dandelion sign). Neurophysiologic examinations were unremarkable.
Discussion: EBF3-related syndrome has been so far described as a neurodevelopmental condition with dysmorphic traits, with limited emphasis on the neurologic features; we highlight the predominant neurologic involvement of these patients, which can be explained at least in part by the underlying cerebellar malformation. We therefore propose that EBF3-related syndrome should be classified and treated as a congenital, nonprogressive ataxia.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


