MethylScore, a pipeline for accurate and context-aware identification of differentially methylated regions from population-scale plant whole-genome bisulfite sequencing data.
Patrick Hüther, Jörg Hagmann, Adam Nunn, Ioanna Kakoulidou, Rahul Pisupati, David Langenberger, Detlef Weigel, Frank Johannes, Sebastian J Schultheiss, Claude Becker
{"title":"MethylScore, a pipeline for accurate and context-aware identification of differentially methylated regions from population-scale plant whole-genome bisulfite sequencing data.","authors":"Patrick Hüther, Jörg Hagmann, Adam Nunn, Ioanna Kakoulidou, Rahul Pisupati, David Langenberger, Detlef Weigel, Frank Johannes, Sebastian J Schultheiss, Claude Becker","doi":"10.1017/qpb.2022.14","DOIUrl":null,"url":null,"abstract":"<p><p>Whole-genome bisulfite sequencing (WGBS) is the standard method for profiling DNA methylation at single-nucleotide resolution. Different tools have been developed to extract differentially methylated regions (DMRs), often built upon assumptions from mammalian data. Here, we present MethylScore, a pipeline to analyse WGBS data and to account for the substantially more complex and variable nature of plant DNA methylation. MethylScore uses an unsupervised machine learning approach to segment the genome by classification into states of high and low methylation. It processes data from genomic alignments to DMR output and is designed to be usable by novice and expert users alike. We show how MethylScore can identify DMRs from hundreds of samples and how its data-driven approach can stratify associated samples without prior information. We identify DMRs in the <i>A. thaliana</i> 1,001 Genomes dataset to unveil known and unknown genotype-epigenotype associations .</p>","PeriodicalId":20825,"journal":{"name":"Quantitative Plant Biology","volume":"3 ","pages":"e19"},"PeriodicalIF":0.0000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10095865/pdf/","citationCount":"6","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Quantitative Plant Biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1017/qpb.2022.14","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 6
Abstract
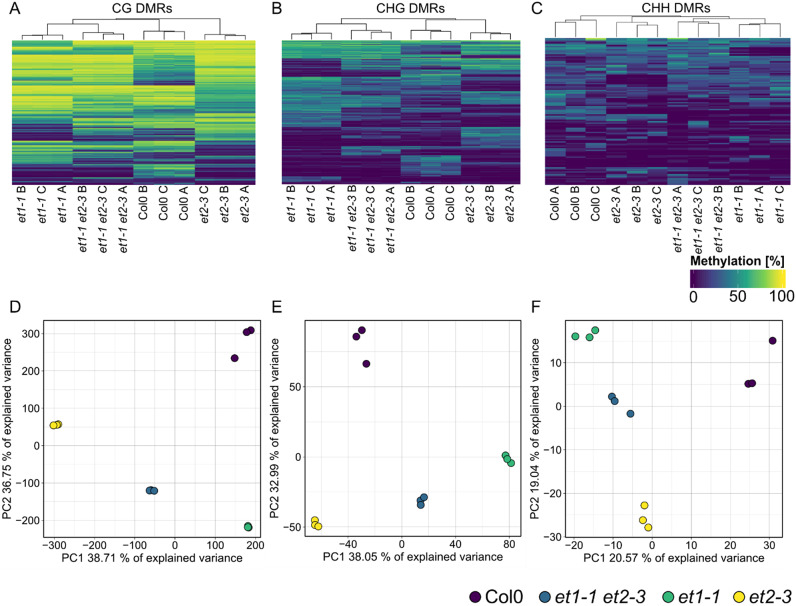
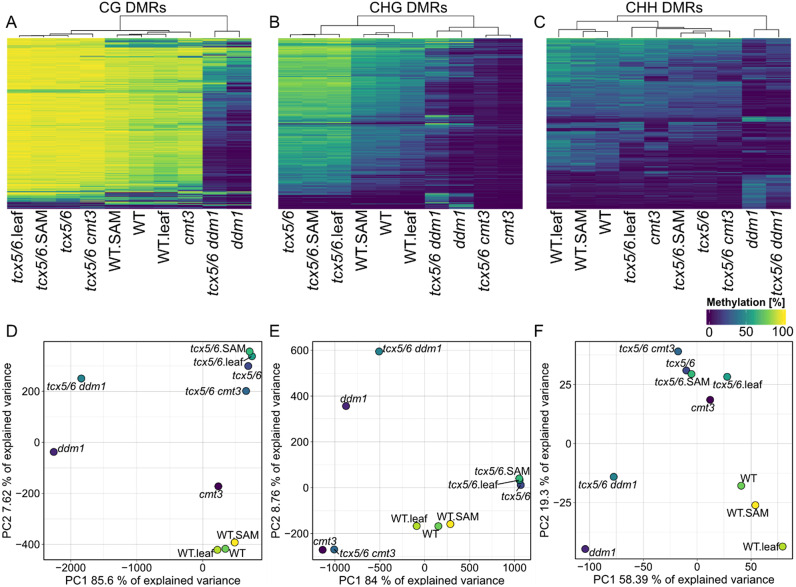
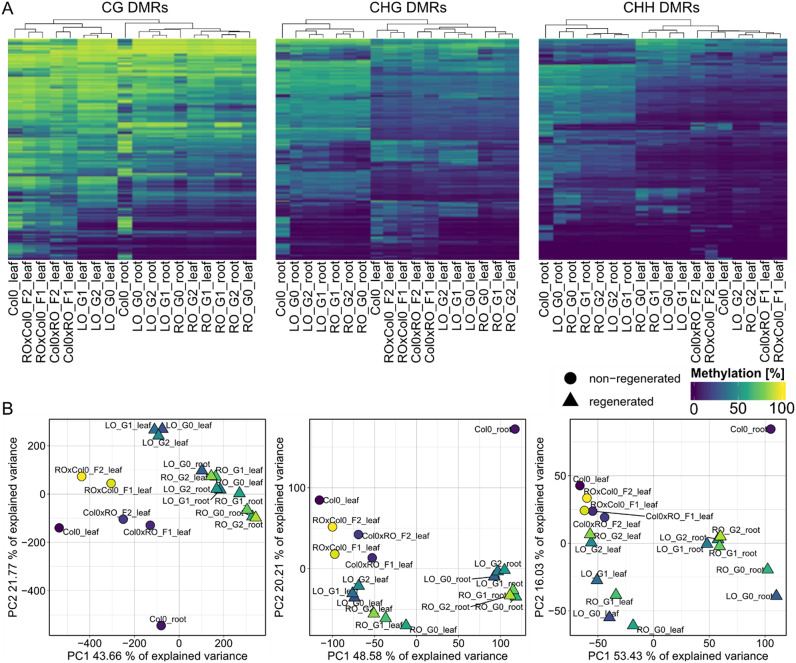
Whole-genome bisulfite sequencing (WGBS) is the standard method for profiling DNA methylation at single-nucleotide resolution. Different tools have been developed to extract differentially methylated regions (DMRs), often built upon assumptions from mammalian data. Here, we present MethylScore, a pipeline to analyse WGBS data and to account for the substantially more complex and variable nature of plant DNA methylation. MethylScore uses an unsupervised machine learning approach to segment the genome by classification into states of high and low methylation. It processes data from genomic alignments to DMR output and is designed to be usable by novice and expert users alike. We show how MethylScore can identify DMRs from hundreds of samples and how its data-driven approach can stratify associated samples without prior information. We identify DMRs in the A. thaliana 1,001 Genomes dataset to unveil known and unknown genotype-epigenotype associations .

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


