Expanding the neuroimaging findings of guanidinoacetate methyltransferase deficiency in an Iranian girl with a homozygous frameshift variant in the GAMT.
Seyedeh Atiyeh Afjei, Mohammad Farid Mohammadi, Elham Pourbakhtyaran, Homa Ghabeli, Mahmoud Reza Ashrafi, Roya Haghighi, Maryam Rasulinezhad, Neda Pak, Ali Reza Tavasoli, Morteza Heidari
{"title":"Expanding the neuroimaging findings of guanidinoacetate methyltransferase deficiency in an Iranian girl with a homozygous frameshift variant in the GAMT.","authors":"Seyedeh Atiyeh Afjei, Mohammad Farid Mohammadi, Elham Pourbakhtyaran, Homa Ghabeli, Mahmoud Reza Ashrafi, Roya Haghighi, Maryam Rasulinezhad, Neda Pak, Ali Reza Tavasoli, Morteza Heidari","doi":"10.1007/s10048-022-00708-2","DOIUrl":null,"url":null,"abstract":"<p><p>Guanidinoacetate methyltransferase deficiency (GAMTD) is a treatable neurodevelopmental disorder with normal or nonspecific imaging findings. Here, we reported a 14-month-old girl with GAMTD and novel findings on brain magnetic resonance imaging (MRI).A 14-month-old female patient was referred to Myelin Disorders Clinic due to onset of seizures and developmental regression following routine vaccination at 4 months of age. Brain MRI, prior to initiation of treatment, showed high signal intensity in T2-weighted imaging in bilateral thalami, globus pallidus, subthalamic nuclei, substantia nigra, dentate nuclei, central tegmental tracts in the brainstem, and posterior periventricular white matter which was masquerading for mitochondrial leukodystrophy. Basic metabolic tests were normal except for low urine creatinine; however, exome sequencing identified a homozygous frameshift deletion variant [NM_000156: c.491del; (p.Gly164AlafsTer14)] in the GAMT. Biallelic pathogenic or likely pathogenic variants cause GAMTD. We confirmed the homozygous state for this variant in the proband, as well as the heterozygote state in the parents by Sanger sequencing.MRI features in GAMTD can mimic mitochondrial leukodystrophy. Pediatric neurologists should be aware of variable MRI findings in GAMTD since they would be misleading to other diagnoses.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":"24 2","pages":"67-78"},"PeriodicalIF":1.6000,"publicationDate":"2023-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-022-00708-2","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
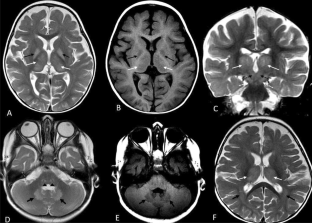
Guanidinoacetate methyltransferase deficiency (GAMTD) is a treatable neurodevelopmental disorder with normal or nonspecific imaging findings. Here, we reported a 14-month-old girl with GAMTD and novel findings on brain magnetic resonance imaging (MRI).A 14-month-old female patient was referred to Myelin Disorders Clinic due to onset of seizures and developmental regression following routine vaccination at 4 months of age. Brain MRI, prior to initiation of treatment, showed high signal intensity in T2-weighted imaging in bilateral thalami, globus pallidus, subthalamic nuclei, substantia nigra, dentate nuclei, central tegmental tracts in the brainstem, and posterior periventricular white matter which was masquerading for mitochondrial leukodystrophy. Basic metabolic tests were normal except for low urine creatinine; however, exome sequencing identified a homozygous frameshift deletion variant [NM_000156: c.491del; (p.Gly164AlafsTer14)] in the GAMT. Biallelic pathogenic or likely pathogenic variants cause GAMTD. We confirmed the homozygous state for this variant in the proband, as well as the heterozygote state in the parents by Sanger sequencing.MRI features in GAMTD can mimic mitochondrial leukodystrophy. Pediatric neurologists should be aware of variable MRI findings in GAMTD since they would be misleading to other diagnoses.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


