{"title":"Genotype-phenotype correlation and natural history study of dysferlinopathy: a single-centre experience from India.","authors":"Saraswati Nashi, Kiran Polavarapu, Mainak Bardhan, Ram Murthy Anjanappa, Veeramani Preethish-Kumar, Seena Vengalil, Hansashree Padmanabha, Thenral S Geetha, P V Prathyusha, Vedam Ramprasad, Aditi Joshi, Tanushree Chawla, Gopikirshnan Unnikrishnan, Pooja Sharma, Akshata Huddar, Bharathram Uppilli, Abel Thomas, Dipti Baskar, Susi Mathew, Deepak Menon, Gautham Arunachal, Mohammed Faruq, Kumarasamy Thangaraj, Atchayaram Nalini","doi":"10.1007/s10048-022-00707-3","DOIUrl":null,"url":null,"abstract":"<p><p>Dysferlinopathies are a group of limb-girdle muscular dystrophies causing significant disability in the young population. There is a need for studies on large cohorts to describe the clinical, genotypic and natural history in our subcontinent. To describe and correlate the clinical, genetic profile and natural history of genetically confirmed dysferlinopathies. We analysed a retrospective cohort of patients with dysferlinopathy from a single quaternary care centre in India. A total of 124 patients with dysferlinopathy were included (40 females). Median age at onset and duration of illness were 21 years (range, 13-50) and 48 months (range, 8-288), respectively. The average follow-up period was 60 months (range, 12-288). Fifty-one percent had LGMD pattern of weakness at onset; 23.4% each had Miyoshi and proximo-distal type while isolated hyperCKemia was noted in 1.6%. About 60% were born to consanguineous parents and 26.6% had family history of similar illness. Twenty-three patients (18.6%) lost ambulation at follow-up; the median time to loss of independent ambulation was 120 months (range, 72-264). Single-nucleotide variants (SNVs) constituted 78.2% of patients; INDELs 14.5% and 7.3% had both SNVs and INDELs. Earlier age at onset was noted with SNVs. There was no correlation between the other clinical parameters and ambulatory status with the genotype. Thirty-seven (45.7%) novel pathogenic/likely pathogenic (P/LP) variants were identified out of a total of 81 variations. The c.3191G > A variant was the most recurrent mutation. Our cohort constitutes a clinically and genetically heterogeneous group of dysferlinopathies. There is no significant correlation between the clinico-genetic profile and the ambulatory status.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":"24 1","pages":"43-53"},"PeriodicalIF":1.6000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-022-00707-3","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
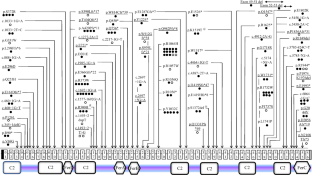
Dysferlinopathies are a group of limb-girdle muscular dystrophies causing significant disability in the young population. There is a need for studies on large cohorts to describe the clinical, genotypic and natural history in our subcontinent. To describe and correlate the clinical, genetic profile and natural history of genetically confirmed dysferlinopathies. We analysed a retrospective cohort of patients with dysferlinopathy from a single quaternary care centre in India. A total of 124 patients with dysferlinopathy were included (40 females). Median age at onset and duration of illness were 21 years (range, 13-50) and 48 months (range, 8-288), respectively. The average follow-up period was 60 months (range, 12-288). Fifty-one percent had LGMD pattern of weakness at onset; 23.4% each had Miyoshi and proximo-distal type while isolated hyperCKemia was noted in 1.6%. About 60% were born to consanguineous parents and 26.6% had family history of similar illness. Twenty-three patients (18.6%) lost ambulation at follow-up; the median time to loss of independent ambulation was 120 months (range, 72-264). Single-nucleotide variants (SNVs) constituted 78.2% of patients; INDELs 14.5% and 7.3% had both SNVs and INDELs. Earlier age at onset was noted with SNVs. There was no correlation between the other clinical parameters and ambulatory status with the genotype. Thirty-seven (45.7%) novel pathogenic/likely pathogenic (P/LP) variants were identified out of a total of 81 variations. The c.3191G > A variant was the most recurrent mutation. Our cohort constitutes a clinically and genetically heterogeneous group of dysferlinopathies. There is no significant correlation between the clinico-genetic profile and the ambulatory status.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


