Birute Burnyte, Ramune Vilimiene, Kristina Grigalioniene, Irina Adomaitiene, Algirdas Utkus
{"title":"Cerebellar Ataxia and Peripheral Neuropathy in a Family With <i>PNPLA8</i>-Associated Disease.","authors":"Birute Burnyte, Ramune Vilimiene, Kristina Grigalioniene, Irina Adomaitiene, Algirdas Utkus","doi":"10.1212/NXG.0000000000200068","DOIUrl":null,"url":null,"abstract":"<p><strong>Objectives: </strong>To describe clinical and genetic findings in 2 siblings with slowly progressive ataxia.</p><p><strong>Methods: </strong>We studied 2 adult siblings through detailed physical and instrumental examinations. Whole-exome sequencing was used to identify an underlying genetic cause.</p><p><strong>Results: </strong>Both siblings presented with adolescence-onset ataxia, progressive sensorimotor polyneuropathy, and preserved cognition over time. The onset of symptoms was between 10 and 14 years of age. A brain MRI demonstrated mild cerebellar atrophy in the older brother at age 45 years. Exome sequencing revealed compound heterozygous loss-of-function variants c.2269del (p.(Thr757GlnfsTer10)) and c.2275_2276del (p.(Leu759AlafsTer4)) in <i>PNPLA8</i>. The novel variant c.2269del results in frameshift with a premature stop codon p.(Thr757GlnfsTer10) and loss of normal enzyme function.</p><p><strong>Discussion: </strong>Our findings support the theory that biallelic loss-of-function <i>PNPLA8</i> variants are involved in neurodegenerative mitochondrial disease. Compared with patients previously described, these patients' phenotype may be interpreted as a milder phenotype associated with a slight progression of ataxia throughout adulthood.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"9 3","pages":"e200068"},"PeriodicalIF":3.0000,"publicationDate":"2023-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/0e/9f/NXG-2023-000011.PMC10088641.pdf","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200068","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 2
Abstract
Objectives: To describe clinical and genetic findings in 2 siblings with slowly progressive ataxia.
Methods: We studied 2 adult siblings through detailed physical and instrumental examinations. Whole-exome sequencing was used to identify an underlying genetic cause.
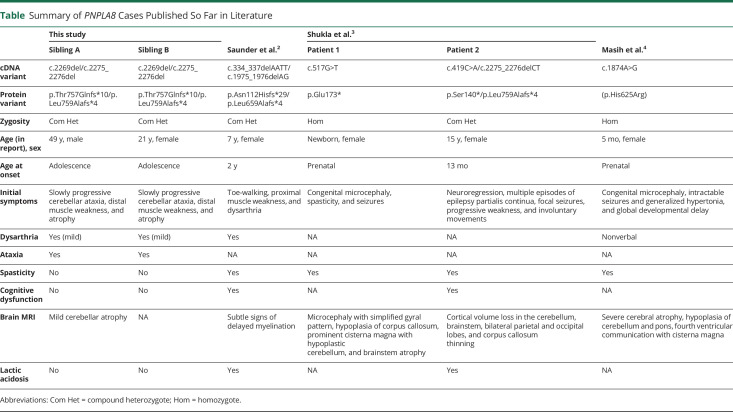
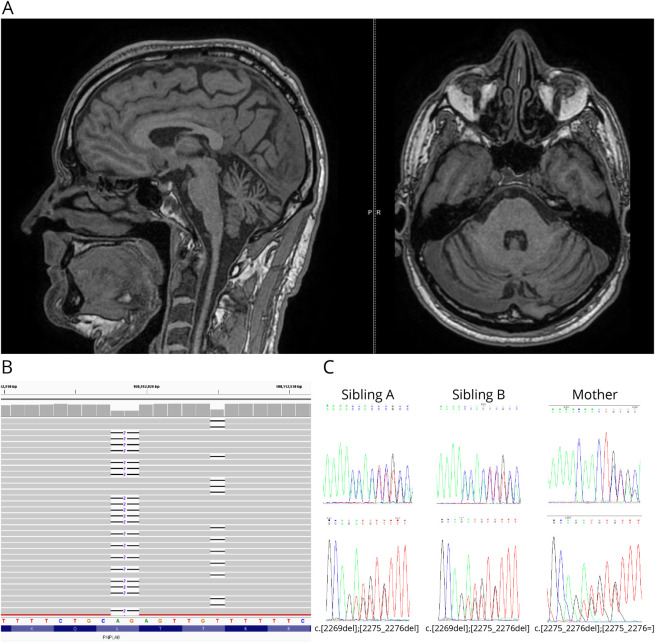
Results: Both siblings presented with adolescence-onset ataxia, progressive sensorimotor polyneuropathy, and preserved cognition over time. The onset of symptoms was between 10 and 14 years of age. A brain MRI demonstrated mild cerebellar atrophy in the older brother at age 45 years. Exome sequencing revealed compound heterozygous loss-of-function variants c.2269del (p.(Thr757GlnfsTer10)) and c.2275_2276del (p.(Leu759AlafsTer4)) in PNPLA8. The novel variant c.2269del results in frameshift with a premature stop codon p.(Thr757GlnfsTer10) and loss of normal enzyme function.
Discussion: Our findings support the theory that biallelic loss-of-function PNPLA8 variants are involved in neurodegenerative mitochondrial disease. Compared with patients previously described, these patients' phenotype may be interpreted as a milder phenotype associated with a slight progression of ataxia throughout adulthood.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


