Chunwang Li, Penghui Liu, Weilin Huang, Haojie Wang, Ke Ma, Lingyun Zhuo, Yaqing Kang, Qiu He, Yuanxiang Lin, Dezhi Kang, Fuxin Lin
{"title":"A novel KRIT1/CCM1 mutation accompanied by a NOTCH3 mutation in a Chinese family with multiple cerebral cavernous malformations.","authors":"Chunwang Li, Penghui Liu, Weilin Huang, Haojie Wang, Ke Ma, Lingyun Zhuo, Yaqing Kang, Qiu He, Yuanxiang Lin, Dezhi Kang, Fuxin Lin","doi":"10.1007/s10048-023-00714-y","DOIUrl":null,"url":null,"abstract":"<p><p>Family cerebral cavernous malformations (FCCMs) are mainly inherited through the mutation of classical CCM genes, including CCM1/KRIT1, CCM2/MGC4607, and CCM3/PDCD10. FCCMs can cause severe clinical symptoms, including epileptic seizures, intracranial hemorrhage (ICH), or functional neurological deficits (FNDs). In this study, we reported a novel mutation in KRIT1 accompanied by a NOTCH3 mutation in a Chinese family. This family consists of 8 members, 4 of whom had been diagnosed with CCMs using cerebral MRI (T1WI, T2WI, SWI). The proband (II-2) and her daughter (III-4) had intracerebral hemorrhage and refractory epilepsy, respectively. Based on whole-exome sequencing (WES) data and bioinformatics analysis from 4 patients with multiple CCMs and 2 normal first-degree relatives, a novel KRIT1 mutation, NG_012964.1 (NM_194456.1): c.1255-1G > T (splice-3), in intron 13 was considered a pathogenic gene in this family. Furthermore, based on 2 severe and 2 mild CCM patients, we found an SNV missense mutation, NG_009819.1 (NM_000435.2): c.1630C > T (p.R544C), in NOTCH3. Finally, the KRIT1 and NOTCH3 mutations were validated in 8 members using Sanger sequencing. This study revealed a novel KRIT1 mutation, NG_012964.1 (NM_194456.1): c.1255-1G > T (splice-3), in a Chinese CCM family, which had not been reported previously. Moreover, the NOTCH3 mutation NG_009819.1 (NM_000435.2): c.1630C > T (p.R544C) might be a second hit and associated with the progression of CCM lesions and severe clinical symptoms.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":"24 2","pages":"137-146"},"PeriodicalIF":1.2000,"publicationDate":"2023-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00714-y","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
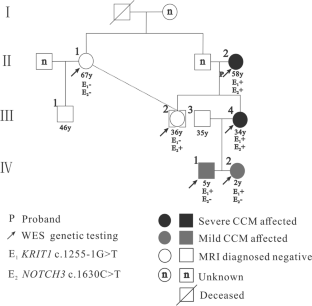
Family cerebral cavernous malformations (FCCMs) are mainly inherited through the mutation of classical CCM genes, including CCM1/KRIT1, CCM2/MGC4607, and CCM3/PDCD10. FCCMs can cause severe clinical symptoms, including epileptic seizures, intracranial hemorrhage (ICH), or functional neurological deficits (FNDs). In this study, we reported a novel mutation in KRIT1 accompanied by a NOTCH3 mutation in a Chinese family. This family consists of 8 members, 4 of whom had been diagnosed with CCMs using cerebral MRI (T1WI, T2WI, SWI). The proband (II-2) and her daughter (III-4) had intracerebral hemorrhage and refractory epilepsy, respectively. Based on whole-exome sequencing (WES) data and bioinformatics analysis from 4 patients with multiple CCMs and 2 normal first-degree relatives, a novel KRIT1 mutation, NG_012964.1 (NM_194456.1): c.1255-1G > T (splice-3), in intron 13 was considered a pathogenic gene in this family. Furthermore, based on 2 severe and 2 mild CCM patients, we found an SNV missense mutation, NG_009819.1 (NM_000435.2): c.1630C > T (p.R544C), in NOTCH3. Finally, the KRIT1 and NOTCH3 mutations were validated in 8 members using Sanger sequencing. This study revealed a novel KRIT1 mutation, NG_012964.1 (NM_194456.1): c.1255-1G > T (splice-3), in a Chinese CCM family, which had not been reported previously. Moreover, the NOTCH3 mutation NG_009819.1 (NM_000435.2): c.1630C > T (p.R544C) might be a second hit and associated with the progression of CCM lesions and severe clinical symptoms.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


