Haitham Gabr, Juan Carlos Rivera-Mulia, David M Gilbert, Tamer Kahveci
{"title":"Computing interaction probabilities in signaling networks.","authors":"Haitham Gabr, Juan Carlos Rivera-Mulia, David M Gilbert, Tamer Kahveci","doi":"10.1186/s13637-015-0031-8","DOIUrl":null,"url":null,"abstract":"<p><p>Biological networks inherently have uncertain topologies. This arises from many factors. For instance, interactions between molecules may or may not take place under varying conditions. Genetic or epigenetic mutations may also alter biological processes like transcription or translation. This uncertainty is often modeled by associating each interaction with a probability value. Studying biological networks under this probabilistic model has already been shown to yield accurate and insightful analysis of interaction data. However, the problem of assigning accurate probability values to interactions remains unresolved. In this paper, we present a novel method for computing interaction probabilities in signaling networks based on transcription levels of genes. The transcription levels define the signal reachability probability between membrane receptors and transcription factors. Our method computes the interaction probabilities that minimize the gap between the observed and the computed signal reachability probabilities. We evaluate our method on four signaling networks from the Kyoto Encyclopedia of Genes and Genomes (KEGG). For each network, we compute its edge probabilities using the gene expression profiles for seven major leukemia subtypes. We use these values to analyze how the stress induced by different leukemia subtypes affects signaling interactions.</p>","PeriodicalId":101453,"journal":{"name":"EURASIP journal on bioinformatics & systems biology","volume":"2015 1","pages":"10"},"PeriodicalIF":0.0000,"publicationDate":"2015-11-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13637-015-0031-8","citationCount":"5","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"EURASIP journal on bioinformatics & systems biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13637-015-0031-8","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2015/12/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 5
Abstract
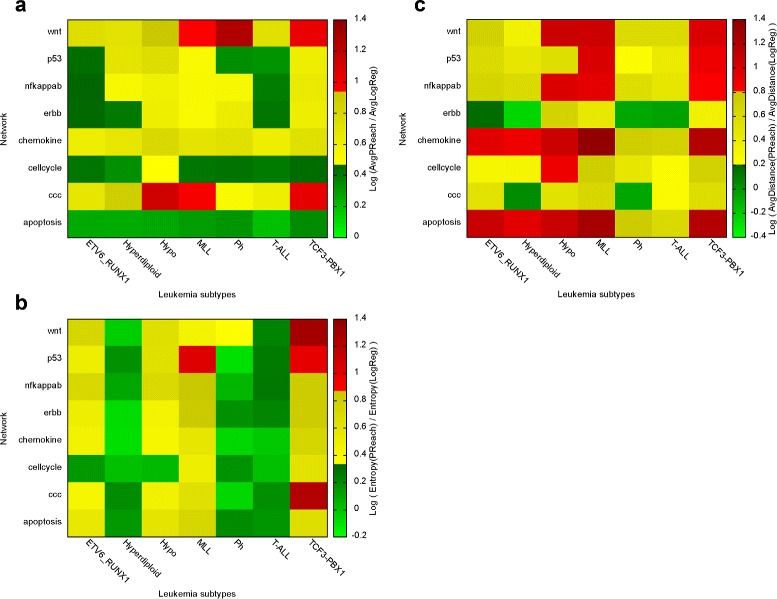
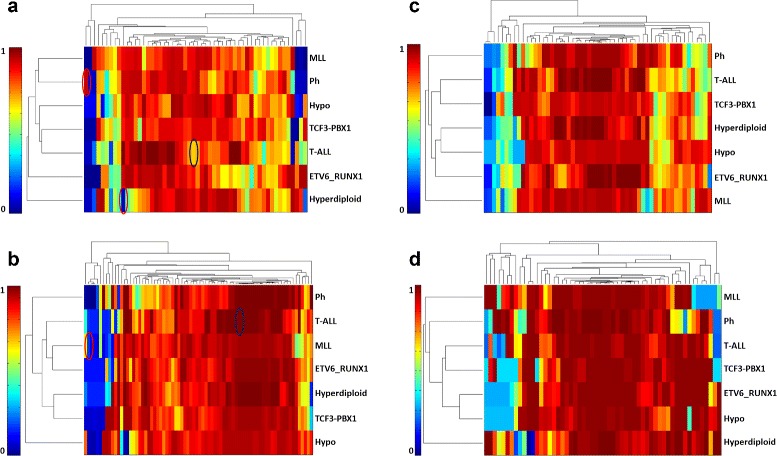
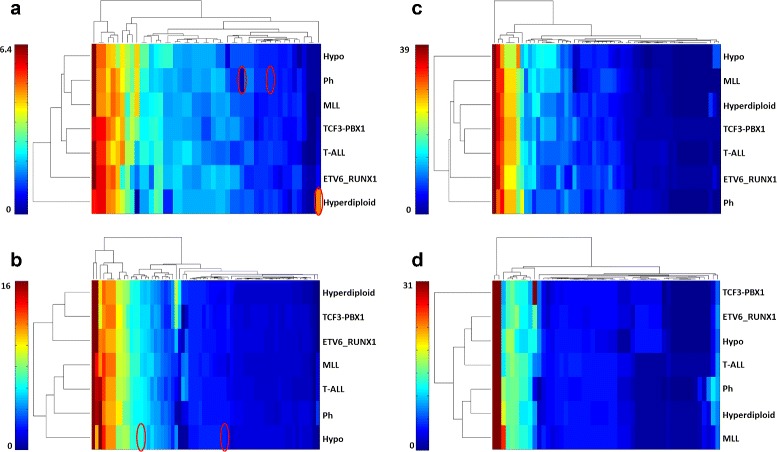
Biological networks inherently have uncertain topologies. This arises from many factors. For instance, interactions between molecules may or may not take place under varying conditions. Genetic or epigenetic mutations may also alter biological processes like transcription or translation. This uncertainty is often modeled by associating each interaction with a probability value. Studying biological networks under this probabilistic model has already been shown to yield accurate and insightful analysis of interaction data. However, the problem of assigning accurate probability values to interactions remains unresolved. In this paper, we present a novel method for computing interaction probabilities in signaling networks based on transcription levels of genes. The transcription levels define the signal reachability probability between membrane receptors and transcription factors. Our method computes the interaction probabilities that minimize the gap between the observed and the computed signal reachability probabilities. We evaluate our method on four signaling networks from the Kyoto Encyclopedia of Genes and Genomes (KEGG). For each network, we compute its edge probabilities using the gene expression profiles for seven major leukemia subtypes. We use these values to analyze how the stress induced by different leukemia subtypes affects signaling interactions.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


