Angel Albavera-Mata, Richard G. Hennig* and S. B. Trickey*,
{"title":"Transition Temperature for Spin-Crossover Materials with the Mean Value Ensemble Hubbard-U Correction","authors":"Angel Albavera-Mata, Richard G. Hennig* and S. B. Trickey*, ","doi":"10.1021/acs.jpca.3c03520","DOIUrl":null,"url":null,"abstract":"<p >Calculation of transition temperatures <i>T</i><sub>1/2</sub> for thermally driven spin-crossover in condensed phases is challenging, even with sophisticated state-of-the-art density functional approximations. The first issue is the accuracy of the adiabatic crossover energy difference <i>ΔE</i><sub>HL</sub> between the low- and high-spin states of the bistable metal–organic complexes. The other is the proper inclusion of entropic contributions to the Gibbs free energy from the electronic and vibrational degrees of freedom. We discuss the effects of treatments of both contributions upon the calculation of thermochemical properties for a set of 20 spin-crossover materials using a Hubbard-<i>U</i> correction obtained from a reference ensemble spin-state. The <i>U</i> values obtained from a simplest bimolecular representation may overcorrect, somewhat, the <i>ΔE</i><sub>HL</sub> values, hence giving somewhat excessive reduction of the <i>T</i><sub>1/2</sub> results with respect to their <i>U</i> = 0 values in the crystalline phase. We discuss the origins of the discrepancies by analyzing different sources of uncertainties. By use of a first-coordination-sphere approximation and the assumption that vibrational contributions from the outermost atoms in a metal–organic complex are similar in both low- and high-spin states, we achieve <i>T</i><sub>1/2</sub> results with the low-cost, widely used PBE generalized gradient density functional approximation comparable to those from the more costly, more sophisticated r<sup>2</sup>SCAN meta-generalized gradient approximation. The procedure is promising for use in high-throughput materials screening, because it combines rather low computational effort requirements with freedom from user manipulation of parameters.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"127 36","pages":"7646–7654"},"PeriodicalIF":2.7000,"publicationDate":"2023-09-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.3c03520","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
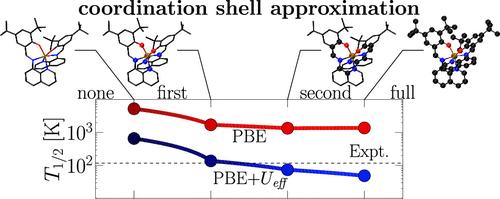
Calculation of transition temperatures T1/2 for thermally driven spin-crossover in condensed phases is challenging, even with sophisticated state-of-the-art density functional approximations. The first issue is the accuracy of the adiabatic crossover energy difference ΔEHL between the low- and high-spin states of the bistable metal–organic complexes. The other is the proper inclusion of entropic contributions to the Gibbs free energy from the electronic and vibrational degrees of freedom. We discuss the effects of treatments of both contributions upon the calculation of thermochemical properties for a set of 20 spin-crossover materials using a Hubbard-U correction obtained from a reference ensemble spin-state. The U values obtained from a simplest bimolecular representation may overcorrect, somewhat, the ΔEHL values, hence giving somewhat excessive reduction of the T1/2 results with respect to their U = 0 values in the crystalline phase. We discuss the origins of the discrepancies by analyzing different sources of uncertainties. By use of a first-coordination-sphere approximation and the assumption that vibrational contributions from the outermost atoms in a metal–organic complex are similar in both low- and high-spin states, we achieve T1/2 results with the low-cost, widely used PBE generalized gradient density functional approximation comparable to those from the more costly, more sophisticated r2SCAN meta-generalized gradient approximation. The procedure is promising for use in high-throughput materials screening, because it combines rather low computational effort requirements with freedom from user manipulation of parameters.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


