{"title":"Two novel cases of biallelic SMPD4 variants with brain structural abnormalities.","authors":"Shintaro Aoki, Kazuki Watanabe, Mitsuhiro Kato, Yukihiko Konishi, Kazuo Kubota, Emiko Kobayashi, Mitsuko Nakashima, Hirotomo Saitsu","doi":"10.1007/s10048-023-00737-5","DOIUrl":null,"url":null,"abstract":"<p><p>Sphingomyelin phosphodiesterase 4 (SMPD4) encodes a member of the Mg<sup>2+</sup>-dependent, neutral sphingomyelinase family that catalyzes the hydrolysis of the phosphodiester bond of sphingomyelin to form phosphorylcholine and ceramide. Recent studies have revealed that biallelic loss-of-function variants of SMPD4 cause syndromic neurodevelopmental disorders characterized by microcephaly, congenital arthrogryposis, and structural brain anomalies. In this study, three novel loss-of-function SMPD4 variants were identified using exome sequencing (ES) in two independent patients with developmental delays, microcephaly, seizures, and brain structural abnormalities. Patient 1 had a homozygous c.740_741del, p.(Val247Glufs*21) variant and showed profound intellectual disability, hepatomegaly, a simplified gyral pattern, and a thin corpus callosum without congenital dysmorphic features. Patient 2 had a compound heterozygous nonsense c.2124_2125del, p.(Phe709*) variant and splice site c.1188+2dup variant. RNA analysis revealed that the c.1188+2dup variant caused exon 13 skipping, leading to a frameshift (p.Ala406Ser*6). In vitro transcription analysis using minigene system suggested that mRNA transcribed from mutant allele may be degraded by nonsense-mediated mRNA decay system. He exhibited diverse manifestations, including growth defects, muscle hypotonia, respiratory distress, arthrogryposis, insulin-dependent diabetes mellitus, sensorineural hearing loss, facial dysmorphism, and various brain abnormalities, including cerebral atrophy, hypomyelination, and cerebellar hypoplasia. Here, we review previous literatures and discuss the phenotypic diversity of SMPD4-related disorders.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":" ","pages":"3-11"},"PeriodicalIF":1.2000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00737-5","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/10/26 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
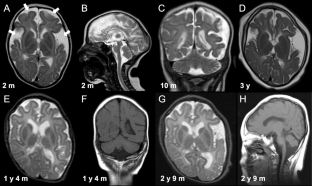
Sphingomyelin phosphodiesterase 4 (SMPD4) encodes a member of the Mg2+-dependent, neutral sphingomyelinase family that catalyzes the hydrolysis of the phosphodiester bond of sphingomyelin to form phosphorylcholine and ceramide. Recent studies have revealed that biallelic loss-of-function variants of SMPD4 cause syndromic neurodevelopmental disorders characterized by microcephaly, congenital arthrogryposis, and structural brain anomalies. In this study, three novel loss-of-function SMPD4 variants were identified using exome sequencing (ES) in two independent patients with developmental delays, microcephaly, seizures, and brain structural abnormalities. Patient 1 had a homozygous c.740_741del, p.(Val247Glufs*21) variant and showed profound intellectual disability, hepatomegaly, a simplified gyral pattern, and a thin corpus callosum without congenital dysmorphic features. Patient 2 had a compound heterozygous nonsense c.2124_2125del, p.(Phe709*) variant and splice site c.1188+2dup variant. RNA analysis revealed that the c.1188+2dup variant caused exon 13 skipping, leading to a frameshift (p.Ala406Ser*6). In vitro transcription analysis using minigene system suggested that mRNA transcribed from mutant allele may be degraded by nonsense-mediated mRNA decay system. He exhibited diverse manifestations, including growth defects, muscle hypotonia, respiratory distress, arthrogryposis, insulin-dependent diabetes mellitus, sensorineural hearing loss, facial dysmorphism, and various brain abnormalities, including cerebral atrophy, hypomyelination, and cerebellar hypoplasia. Here, we review previous literatures and discuss the phenotypic diversity of SMPD4-related disorders.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


