{"title":"Co and Ni single sites on the (111)n surface of γ-Al2O3 – a periodic boundary DFT study†","authors":"Jiande Gu, Jing Wang and Jerzy Leszczynski","doi":"10.1039/D2IM00039C","DOIUrl":null,"url":null,"abstract":"<p>The influences of increasing the number of d-electrons in the single metal (Fe-like) substituted (111)<small><sub><em>n</em></sub></small> surface of γ-Al<small><sub>2</sub></small>O<small><sub>3</sub></small> on its possible catalytic effects were explored. The energetic properties, local structures, and in-site electron configurations of the most active tri-coordinated Co and Ni single-site (111)<small><sub><em>n</em></sub></small> surface of γ-Al<small><sub>2</sub></small>O<small><sub>3</sub></small> have been studied using the density functional theory (DFT) approach under periodic boundary conditions. The replacement of Al by a Co or Ni atom on the I position of the (111)<small><sub><em>n</em></sub></small> surface leads to significant elongations of metal–O distances. The energy released from the substitution process on the Al<small><sub>I</sub></small> site of the (111)<small><sub><em>n</em></sub></small> surface follows the sequence Ni<small><sub>I</sub></small> (164.85 kcal mol<small><sup>−1</sup></small>) > Co<small><sub>I</sub></small> (113.17 kcal mol<small><sup>−1</sup></small>) > Fe<small><sub>I</sub></small> (44.30 kcal mol<small><sup>−1</sup></small>). The triplet and quintet (ground state) of the Co<small><sub>I</sub></small> substituted complex are energy degenerate. Also, the doublet and quartet (ground state) of the Ni<small><sub>I</sub></small> substituted complex have the same stable energy. This energy degeneracy comes from the α–β electron flipping on the p-orbital of the neighboring O that is next to the substituted Co<small><sub>I</sub></small> or Ni<small><sub>I</sub></small> site on the (111)<small><sub><em>n</em></sub></small> surface of γ-Al<small><sub>2</sub></small>O<small><sub>3</sub></small>. Different from the Fe<small><sub>I</sub></small> substituted single-site (111)<small><sub><em>n</em></sub></small> surface, in which the electron configuration of Fe<small><sub>I</sub></small> varies according to its spin-multiplicity state, substituted Ni<small><sub>I</sub></small> has a unique d<small><sup>8</sup></small> electron configuration in all three spin states, and similarly, Co<small><sub>I</sub></small> has a unique d<small><sup>7</sup></small> electron configuration in all three open shell spin states. An increase of the population of d-electrons in the single metal substituted (111)<small><sub><em>n</em></sub></small> surface of γ-Al<small><sub>2</sub></small>O<small><sub>3</sub></small> is likely to provide a more stable electron configuration in the metal catalytic center.</p><p>Keywords: Co substituted surface of γ-Al<small><sub>2</sub></small>O<small><sub>3</sub></small>; Ni substituted surface of γ-Al<small><sub>2</sub></small>O<small><sub>3</sub></small>; (111)<small><sub><em>n</em></sub></small> surface; Periodic boundary DFT approach; Metal catalytic center.</p>","PeriodicalId":29808,"journal":{"name":"Industrial Chemistry & Materials","volume":" 1","pages":" 117-128"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2023/im/d2im00039c?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Industrial Chemistry & Materials","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2023/im/d2im00039c","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
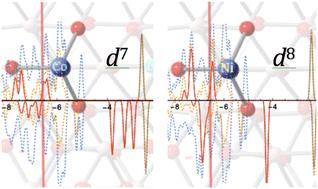
The influences of increasing the number of d-electrons in the single metal (Fe-like) substituted (111)n surface of γ-Al2O3 on its possible catalytic effects were explored. The energetic properties, local structures, and in-site electron configurations of the most active tri-coordinated Co and Ni single-site (111)n surface of γ-Al2O3 have been studied using the density functional theory (DFT) approach under periodic boundary conditions. The replacement of Al by a Co or Ni atom on the I position of the (111)n surface leads to significant elongations of metal–O distances. The energy released from the substitution process on the AlI site of the (111)n surface follows the sequence NiI (164.85 kcal mol−1) > CoI (113.17 kcal mol−1) > FeI (44.30 kcal mol−1). The triplet and quintet (ground state) of the CoI substituted complex are energy degenerate. Also, the doublet and quartet (ground state) of the NiI substituted complex have the same stable energy. This energy degeneracy comes from the α–β electron flipping on the p-orbital of the neighboring O that is next to the substituted CoI or NiI site on the (111)n surface of γ-Al2O3. Different from the FeI substituted single-site (111)n surface, in which the electron configuration of FeI varies according to its spin-multiplicity state, substituted NiI has a unique d8 electron configuration in all three spin states, and similarly, CoI has a unique d7 electron configuration in all three open shell spin states. An increase of the population of d-electrons in the single metal substituted (111)n surface of γ-Al2O3 is likely to provide a more stable electron configuration in the metal catalytic center.
Keywords: Co substituted surface of γ-Al2O3; Ni substituted surface of γ-Al2O3; (111)n surface; Periodic boundary DFT approach; Metal catalytic center.
期刊介绍:
Industrial Chemistry & Materials (ICM) publishes significant innovative research and major technological breakthroughs in all aspects of industrial chemistry and materials, with a particular focus on the important innovation of low-carbon chemical industry, energy and functional materials. By bringing researchers, engineers, and policymakers into one place, research is inspired, challenges are solved and the applications of science and technology are accelerated.
The global editorial and advisory board members are valued experts in the community. With their support, the rigorous editorial practices and dissemination ensures your research is accessible and discoverable on a global scale.
Industrial Chemistry & Materials publishes:
● Communications
● Full papers
● Minireviews
● Reviews
● Perspectives
● Comments

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


