{"title":"Theoretical study of the Meisenheimer and charge-transfer complexes formed upon colorimetric determination of nitroaromatic explosives","authors":"Sergey V. Bondarchuk","doi":"10.1016/j.fpc.2022.09.004","DOIUrl":null,"url":null,"abstract":"<div><p>In this paper, we present a theoretical study of structure and UV-vis spectra of 11 colored complexes of nucleophiles with nitroaromatic energetic materials. Two different schemes were found to be the most suitable for absorption spectra simulation. In the case of covalently bound Meisenheimer complexes, the time-dependent density functional theory (TD-DFT) approach with the TPSS functional was the most accurate. Meanwhile, for intermolecular charge-transfer complexes, the closest spectral pattern was provided by the time-dependent Hartree-Fock (TD-HF) scheme with modified exchange contribution (40%). It has been found that the binding type is determined predominantly by the steric factors and less by the electronic effects of the nucleophile, which was approved by the quantum theory of atoms in molecules (QTAIM) analysis of the formed bond types and nucleophilicity index calculations. For the charge-transfer complex, an appropriate configuration with the intermolecular separation between the local electrophilic and nucleophilic sites (the C1···N distance) of about 3.1 Å, was revealed using both classical molecular dynamics simulations and geometry optimizations in polar continuum. Absorption energies and intensities of the electronic transitions are generally well-reproduced in all 11 cases and demonstrate a local π–π* excitation in the covalently-bound complexes and pure charge transfer in intermolecular system. The applied computational methods allow reproducing of the sample colors with a high degree of similarity, which may find their application for modeling of new reagents with other expected colorimetric characteristics.</p></div>","PeriodicalId":100531,"journal":{"name":"FirePhysChem","volume":"3 2","pages":"Pages 164-172"},"PeriodicalIF":3.6000,"publicationDate":"2023-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"FirePhysChem","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2667134422000487","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 1
Abstract
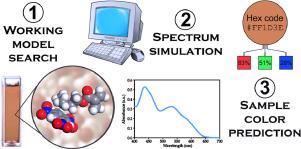
In this paper, we present a theoretical study of structure and UV-vis spectra of 11 colored complexes of nucleophiles with nitroaromatic energetic materials. Two different schemes were found to be the most suitable for absorption spectra simulation. In the case of covalently bound Meisenheimer complexes, the time-dependent density functional theory (TD-DFT) approach with the TPSS functional was the most accurate. Meanwhile, for intermolecular charge-transfer complexes, the closest spectral pattern was provided by the time-dependent Hartree-Fock (TD-HF) scheme with modified exchange contribution (40%). It has been found that the binding type is determined predominantly by the steric factors and less by the electronic effects of the nucleophile, which was approved by the quantum theory of atoms in molecules (QTAIM) analysis of the formed bond types and nucleophilicity index calculations. For the charge-transfer complex, an appropriate configuration with the intermolecular separation between the local electrophilic and nucleophilic sites (the C1···N distance) of about 3.1 Å, was revealed using both classical molecular dynamics simulations and geometry optimizations in polar continuum. Absorption energies and intensities of the electronic transitions are generally well-reproduced in all 11 cases and demonstrate a local π–π* excitation in the covalently-bound complexes and pure charge transfer in intermolecular system. The applied computational methods allow reproducing of the sample colors with a high degree of similarity, which may find their application for modeling of new reagents with other expected colorimetric characteristics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


