Initial unimolecular decomposition of 3,4-bis(3-fluorodinitromethylfuroxan-4-yl) furoxan from quantum mechanics and ReaxFF molecular dynamics simulation
Abstract
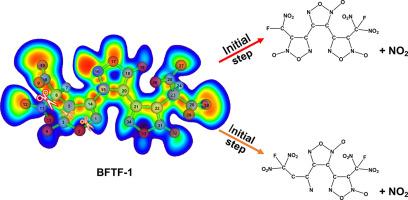
High-energy-density materials (HEDMs) have a wide range of applications in many usages. Recently synthesized 3,4-bis(3-fluorodinitromethylfuroxan-4-yl) furoxan (BFTF-1), composed of furoxan rings and fluorodinitromethyl groups, has shown advanced properties comparing to other existed HEDMs, such as density and enthalpy of formation. Understanding the decomposition mechanism for BFTF-1 could provide insights into future designs of HEDMs, and the initial decompositions are critical steps in the mechanism. In the present study, we employed quantum mechanics calculations and reactive molecular dynamics simulations to explore the initial decomposition steps. The electronic structural analysis and bond dissociation energy calculations suggested that the nitro moieties in the fluorodinitromethyl groups and the furoxan rings could begin the bond breaking process in BFTF-1. The reactive molecular dynamics simulation showed that the increase of the nitro moieties was concurrent with the decrease of BFTF-1, proving the nitro moieties were the first product for the decomposition of BFTF-1. The present study laid the ground for the theoretical understanding of decomposition mechanisms for BFTF-1 and shed light on further designs of advanced HEDMs.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


