{"title":"Exploring computational tools for improved structural design and stability of helical AApeptides","authors":"Tongtong Li , Shenghan Song , Yi He","doi":"10.1016/j.supmat.2023.100038","DOIUrl":null,"url":null,"abstract":"<div><p>Peptidomimetics have garnered increased attention due to their unique structural properties and self-assembly, as well as their promising applications in material sciences and peptidomimetic drug design. N-acetylated-N-aminoethyl amino acid oligomers (AApeptides), a novel class of helical foldamer, have been effectively utilized to imitate protein helices and regulate protein-protein interactions. While the structures of a series of AApeptides have been determined experimentally, the chemical diversity of AApeptides is impeding the advancement of peptide inhibitor development and high-level architecture design through purely experimental methods. Consequently, there's an urgent need for effective computational tools to facilitate the structural design of more complex systems using AApeptides. While a general AMBER force field (GAFF) can be applied to simulate AApeptides, it is crucial to evaluate the accuracy of such a force field and consider alternatives to enhance accuracy. In this study, we employed molecular dynamics simulations (MD) to assess the stability of a helical AApeptide. Our findings indicate that GAFF alone is insufficient to stabilize the helical structure of our AApeptide. We suggest the use of restraints derived from experimentally determined structures to guide the simulation and maintain this helical structure. Although any set of restraint definitions used in this study can simulate helical packing, a minimum of three points per AApeptide building block restraint is necessary to accurately reproduce the hydrogen bonding pattern.</p></div>","PeriodicalId":101187,"journal":{"name":"Supramolecular Materials","volume":"2 ","pages":"Article 100038"},"PeriodicalIF":0.0000,"publicationDate":"2023-08-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Supramolecular Materials","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2667240523000089","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
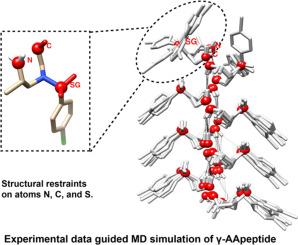
Peptidomimetics have garnered increased attention due to their unique structural properties and self-assembly, as well as their promising applications in material sciences and peptidomimetic drug design. N-acetylated-N-aminoethyl amino acid oligomers (AApeptides), a novel class of helical foldamer, have been effectively utilized to imitate protein helices and regulate protein-protein interactions. While the structures of a series of AApeptides have been determined experimentally, the chemical diversity of AApeptides is impeding the advancement of peptide inhibitor development and high-level architecture design through purely experimental methods. Consequently, there's an urgent need for effective computational tools to facilitate the structural design of more complex systems using AApeptides. While a general AMBER force field (GAFF) can be applied to simulate AApeptides, it is crucial to evaluate the accuracy of such a force field and consider alternatives to enhance accuracy. In this study, we employed molecular dynamics simulations (MD) to assess the stability of a helical AApeptide. Our findings indicate that GAFF alone is insufficient to stabilize the helical structure of our AApeptide. We suggest the use of restraints derived from experimentally determined structures to guide the simulation and maintain this helical structure. Although any set of restraint definitions used in this study can simulate helical packing, a minimum of three points per AApeptide building block restraint is necessary to accurately reproduce the hydrogen bonding pattern.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


