{"title":"Phase transitions and compressibility of alkali-bearing double carbonates at high pressures: a first-principles calculations study","authors":"Bingxu Hou, Shengxuan Huang, Shan Qin","doi":"10.1007/s00269-022-01210-9","DOIUrl":null,"url":null,"abstract":"<div><p>Here, we investigated high-pressure behaviors of four end-members of K-Na-Ca-Mg alkali-bearing double carbonates (K<sub>2</sub>Mg(CO<sub>3</sub>)<sub>2</sub>, K<sub>2</sub>Ca(CO<sub>3</sub>)<sub>2</sub>, Na<sub>2</sub>Mg(CO<sub>3</sub>)<sub>2</sub>, and Na<sub>2</sub>Ca(CO<sub>3</sub>)<sub>2</sub>) using first-principles calculations up to ~ 25 GPa. For K<sub>2</sub>Mg, K<sub>2</sub>Ca, and Na<sub>2</sub>Mg double carbonates, the transitions from rhombohedral structures (<i>R</i> <span>\\(\\stackrel{\\mathrm{-}}{3}\\)</span> <i>m</i> or <i>R</i> <span>\\(\\stackrel{\\mathrm{-}}{3}\\)</span>) to monoclinic (<i>C</i>2/<i>m</i>) or triclinic (<i>P</i> <span>\\(\\stackrel{\\mathrm{-}}{1}\\)</span>) structures are predicted. While for Na<sub>2</sub>Ca(CO<sub>3</sub>)<sub>2</sub>, the <i>P</i>2<sub>1</sub><i>ca</i> structure remains stable across the calculated pressure range. But the high-pressure behavior of Na<sub>2</sub>Ca double carbonate has changed over 8 GPa: the <i>b</i>-axis becomes more compressible than <i>a</i>-axis; [CO<sub>3</sub>] –I groups tilt out of the <i>a</i>-<i>b</i> plane upon compression and reverse the direction of rotation at 8 GPa. The parameters for the equations of state of these minerals and their high-pressure phases were all theoretically determined. The predicted transformation is driven by the differences in the compressibility of structural units. The K<sup>+</sup> and Na<sup>+</sup> coordination polyhedra are more compressible in the structure, compared with the high axial rigidity of C–O bonds in the [CO<sub>3</sub>] triangle along the <i>a-b</i> plane. Our results provide projections of the high-pressure behaviors of trigonal double carbonates, in part by helping to clarify the relation among the average metallic ionic radius (<i>R</i><sub>avg</sub>), the bulk modulus (<i>K</i><sub>0</sub>), and the transition pressure (<i>P</i><sub>T</sub>). The transition pressure (<i>P</i><sub>T</sub>) is anticorrelated to the average metallic ionic radius (<i>R</i><sub>avg</sub>), and a larger <i>R</i><sub>avg</sub> results in a lower bulk modulus (<i>K</i><sub>0</sub>) for the trigonal double carbonates. Furthermore, alkali-bearing double carbonates found as inclusions in the natural diamond may indicate a hydrous parental medium composition and a deeper genesis mechanism.</p></div>","PeriodicalId":20132,"journal":{"name":"Physics and Chemistry of Minerals","volume":"49 8","pages":""},"PeriodicalIF":1.2000,"publicationDate":"2022-08-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physics and Chemistry of Minerals","FirstCategoryId":"89","ListUrlMain":"https://link.springer.com/article/10.1007/s00269-022-01210-9","RegionNum":4,"RegionCategory":"地球科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
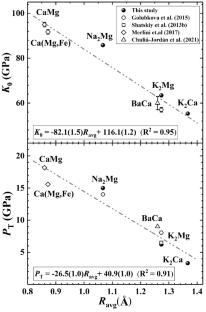
Here, we investigated high-pressure behaviors of four end-members of K-Na-Ca-Mg alkali-bearing double carbonates (K2Mg(CO3)2, K2Ca(CO3)2, Na2Mg(CO3)2, and Na2Ca(CO3)2) using first-principles calculations up to ~ 25 GPa. For K2Mg, K2Ca, and Na2Mg double carbonates, the transitions from rhombohedral structures (R\(\stackrel{\mathrm{-}}{3}\)m or R\(\stackrel{\mathrm{-}}{3}\)) to monoclinic (C2/m) or triclinic (P\(\stackrel{\mathrm{-}}{1}\)) structures are predicted. While for Na2Ca(CO3)2, the P21ca structure remains stable across the calculated pressure range. But the high-pressure behavior of Na2Ca double carbonate has changed over 8 GPa: the b-axis becomes more compressible than a-axis; [CO3] –I groups tilt out of the a-b plane upon compression and reverse the direction of rotation at 8 GPa. The parameters for the equations of state of these minerals and their high-pressure phases were all theoretically determined. The predicted transformation is driven by the differences in the compressibility of structural units. The K+ and Na+ coordination polyhedra are more compressible in the structure, compared with the high axial rigidity of C–O bonds in the [CO3] triangle along the a-b plane. Our results provide projections of the high-pressure behaviors of trigonal double carbonates, in part by helping to clarify the relation among the average metallic ionic radius (Ravg), the bulk modulus (K0), and the transition pressure (PT). The transition pressure (PT) is anticorrelated to the average metallic ionic radius (Ravg), and a larger Ravg results in a lower bulk modulus (K0) for the trigonal double carbonates. Furthermore, alkali-bearing double carbonates found as inclusions in the natural diamond may indicate a hydrous parental medium composition and a deeper genesis mechanism.
期刊介绍:
Physics and Chemistry of Minerals is an international journal devoted to publishing articles and short communications of physical or chemical studies on minerals or solids related to minerals. The aim of the journal is to support competent interdisciplinary work in mineralogy and physics or chemistry. Particular emphasis is placed on applications of modern techniques or new theories and models to interpret atomic structures and physical or chemical properties of minerals. Some subjects of interest are:
-Relationships between atomic structure and crystalline state (structures of various states, crystal energies, crystal growth, thermodynamic studies, phase transformations, solid solution, exsolution phenomena, etc.)
-General solid state spectroscopy (ultraviolet, visible, infrared, Raman, ESCA, luminescence, X-ray, electron paramagnetic resonance, nuclear magnetic resonance, gamma ray resonance, etc.)
-Experimental and theoretical analysis of chemical bonding in minerals (application of crystal field, molecular orbital, band theories, etc.)
-Physical properties (magnetic, mechanical, electric, optical, thermodynamic, etc.)
-Relations between thermal expansion, compressibility, elastic constants, and fundamental properties of atomic structure, particularly as applied to geophysical problems
-Electron microscopy in support of physical and chemical studies
-Computational methods in the study of the structure and properties of minerals
-Mineral surfaces (experimental methods, structure and properties)

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


