Role of solvation model on the stability of oxygenates on Pt(111): A comparison between microsolvation, extended bilayer, and extended metal/water interface
{"title":"Role of solvation model on the stability of oxygenates on Pt(111): A comparison between microsolvation, extended bilayer, and extended metal/water interface","authors":"Giovanni Di Liberto, Livia Giordano","doi":"10.1002/elsa.202100204","DOIUrl":null,"url":null,"abstract":"<p>The activity of catalysts is mainly dictated by the adsorption strength of reaction intermediates at their surfaces. For electrocatalysts in solution, the adsorption strength is not only determined by the intrinsic properties of catalysts and reactants, but also by the solvation energy of reaction intermediates, which is difficult to capture with theoretical methods. Here, we report the impact of different explicit solvation approaches in estimating the stability of oxygenates on the (111) surface of platinum, widely used in oxygen electrocatalysis. We simulate the adsorption of OH, O, and OOH intermediates, relevant for oxygen reduction and oxygen evolution reactions, on Pt(111) with different solvation environments. We apply the static water bilayer model, typically adopted to calculate solvation energies on Pt(111) in computational studies. We then study the trend of solvation energies under different microsolvation environments, by adsorbing the intermediates in presence of an increasing number of water molecules. Last, we use a dynamic approach based on ab-initio molecular dynamics (AIMD) to account for dynamic effects. Our results indicate that the stabilities of oxygenates approach those of the water bilayer when the number of molecules increases from zero to three, but the free energies are affected in a not trivial way by the morphology and size of the water cluster, due to the increased complexity and configurational space. Moreover, static methods imply overcorrected free energies. The adoption of a molecular dynamics approach, based on single-run AIMD simulation of the Pt(111)/H<sub>2</sub>O interface, allows retrieval estimates close to the experimental observation, including dynamic effects, and is highly transferrable. These results suggest that i) when using a microsolvation scheme, it is recommended to include a few water molecules, up to three to resemble the picture of the static bilayer model; ii) dynamic effects are important and can be included with a single-run AIMD scheme.</p>","PeriodicalId":93746,"journal":{"name":"Electrochemical science advances","volume":"4 1","pages":""},"PeriodicalIF":4.1000,"publicationDate":"2023-02-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/elsa.202100204","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Electrochemical science advances","FirstCategoryId":"1085","ListUrlMain":"https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/elsa.202100204","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"ELECTROCHEMISTRY","Score":null,"Total":0}
引用次数: 0
Abstract
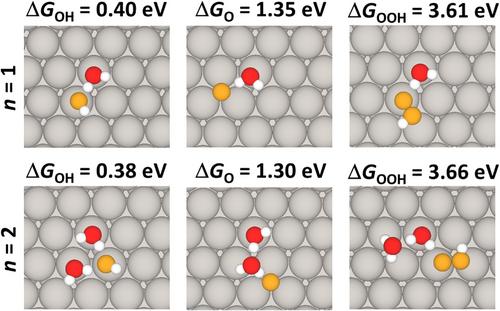
The activity of catalysts is mainly dictated by the adsorption strength of reaction intermediates at their surfaces. For electrocatalysts in solution, the adsorption strength is not only determined by the intrinsic properties of catalysts and reactants, but also by the solvation energy of reaction intermediates, which is difficult to capture with theoretical methods. Here, we report the impact of different explicit solvation approaches in estimating the stability of oxygenates on the (111) surface of platinum, widely used in oxygen electrocatalysis. We simulate the adsorption of OH, O, and OOH intermediates, relevant for oxygen reduction and oxygen evolution reactions, on Pt(111) with different solvation environments. We apply the static water bilayer model, typically adopted to calculate solvation energies on Pt(111) in computational studies. We then study the trend of solvation energies under different microsolvation environments, by adsorbing the intermediates in presence of an increasing number of water molecules. Last, we use a dynamic approach based on ab-initio molecular dynamics (AIMD) to account for dynamic effects. Our results indicate that the stabilities of oxygenates approach those of the water bilayer when the number of molecules increases from zero to three, but the free energies are affected in a not trivial way by the morphology and size of the water cluster, due to the increased complexity and configurational space. Moreover, static methods imply overcorrected free energies. The adoption of a molecular dynamics approach, based on single-run AIMD simulation of the Pt(111)/H2O interface, allows retrieval estimates close to the experimental observation, including dynamic effects, and is highly transferrable. These results suggest that i) when using a microsolvation scheme, it is recommended to include a few water molecules, up to three to resemble the picture of the static bilayer model; ii) dynamic effects are important and can be included with a single-run AIMD scheme.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


