{"title":"Dye Modified Phenylenediamine Oligomers: Theoretical Studies on Drug Binding for Their Potential Application in Drug Sensors","authors":"Ufana Riaz*, and , Syed Marghoob Ashraf, ","doi":"10.1021/acsphyschemau.3c00025","DOIUrl":null,"url":null,"abstract":"<p >The present work reports, for the first time, synthesis of dye incorporated <i>o</i>-phenylenediamine (OBB) with a view to obtain a conjugated oligomer with enhanced functionality. The structure was confirmed by IR studies, while the electronic transitions were confirmed by UV visible studies. The dye modified oligomer showed one order higher fluorescence intensity than the pristine Bismarck Brown (BB) dye. Confocal imaging showed red emission which could be utilized in near infra-red imaging. Density functional theory (DFT) studies were carried out to predict the theoretical properties of the oligomers. The energies of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital orbital were computed to explore how the HOMO energies of the reactants initiated the electronic interactions between them. The interaction energies were correlated to conjugation/hyper conjugation stabilization energies of the natural bond orbitals (NBO) via the DFT method using the B3LYP functional with the 6-311G(d) basis set on Gaussian 09 software. Drug binding was evaluated through simulation of interaction energy, (Δ<i>E</i><sub><i>A</i>–<i>x</i></sub>) with drugs such as captopril, propranolol, thiazide, and fentanyl. The results predicted that the oligomer could be developed into a fentanyl drug sensor.</p>","PeriodicalId":29796,"journal":{"name":"ACS Physical Chemistry Au","volume":"3 6","pages":"521–531"},"PeriodicalIF":3.7000,"publicationDate":"2023-08-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acsphyschemau.3c00025","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Physical Chemistry Au","FirstCategoryId":"1085","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acsphyschemau.3c00025","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
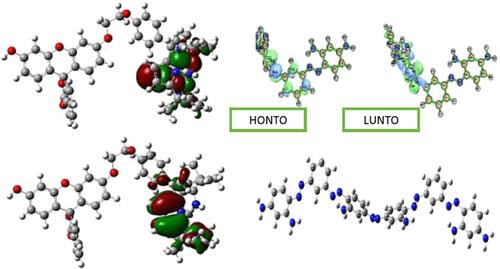
The present work reports, for the first time, synthesis of dye incorporated o-phenylenediamine (OBB) with a view to obtain a conjugated oligomer with enhanced functionality. The structure was confirmed by IR studies, while the electronic transitions were confirmed by UV visible studies. The dye modified oligomer showed one order higher fluorescence intensity than the pristine Bismarck Brown (BB) dye. Confocal imaging showed red emission which could be utilized in near infra-red imaging. Density functional theory (DFT) studies were carried out to predict the theoretical properties of the oligomers. The energies of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital orbital were computed to explore how the HOMO energies of the reactants initiated the electronic interactions between them. The interaction energies were correlated to conjugation/hyper conjugation stabilization energies of the natural bond orbitals (NBO) via the DFT method using the B3LYP functional with the 6-311G(d) basis set on Gaussian 09 software. Drug binding was evaluated through simulation of interaction energy, (ΔEA–x) with drugs such as captopril, propranolol, thiazide, and fentanyl. The results predicted that the oligomer could be developed into a fentanyl drug sensor.
期刊介绍:
ACS Physical Chemistry Au is an open access journal which publishes original fundamental and applied research on all aspects of physical chemistry. The journal publishes new and original experimental computational and theoretical research of interest to physical chemists biophysical chemists chemical physicists physicists material scientists and engineers. An essential criterion for acceptance is that the manuscript provides new physical insight or develops new tools and methods of general interest. Some major topical areas include:Molecules Clusters and Aerosols; Biophysics Biomaterials Liquids and Soft Matter; Energy Materials and Catalysis

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


