Multiomics characterisation of the zoo-housed gorilla gut microbiome reveals bacterial community compositions shifts, fungal cellulose-degrading, and archaeal methanogenic activity.
Isabel M Houtkamp, Martine van Zijll Langhout, Mark Bessem, Walter Pirovano, Remco Kort
{"title":"Multiomics characterisation of the zoo-housed gorilla gut microbiome reveals bacterial community compositions shifts, fungal cellulose-degrading, and archaeal methanogenic activity.","authors":"Isabel M Houtkamp, Martine van Zijll Langhout, Mark Bessem, Walter Pirovano, Remco Kort","doi":"10.1017/gmb.2023.11","DOIUrl":null,"url":null,"abstract":"<p><p>We carried out a comparative analysis between the bacterial microbiota composition of zoo-housed western lowland gorillas and their wild counterparts through 16S rRNA gene amplicon sequencing. In addition, we characterised the carbohydrate-active and methanogenic potential of the zoo-housed gorilla (ZHG) microbiome through shotgun metagenomics and RNA sequencing. The ZHG microbiota showed increased alpha diversity in terms of bacterial species richness and a distinct composition from that of the wild gorilla microbiota, including a loss of abundant fibre-degrading and hydrogenic Chloroflexi. Metagenomic analysis of the CAZyome indicated predominant oligosaccharide-degrading activity, while RNA sequencing revealed diverse cellulase and hemi-cellulase activities in the ZHG gut, contributing to a total of 268 identified carbohydrate-active enzymes. Metatranscriptome analysis revealed a substantial contribution of 38% of the transcripts from anaerobic fungi and archaea to the gorilla microbiome. This activity originates from cellulose-degrading and hydrogenic fungal species belonging to the class Neocallimastigomycetes, as well as from methylotrophic and hydrogenotrophic methanogenic archaea belonging to the classes Thermoplasmata and Methanobacteria, respectively. Our study shows the added value of RNA sequencing in a multiomics approach and highlights the contribution of eukaryotic and archaeal activities to the gut microbiome of gorillas.</p>","PeriodicalId":73187,"journal":{"name":"Gut microbiome (Cambridge, England)","volume":" ","pages":"e12"},"PeriodicalIF":0.0000,"publicationDate":"2023-07-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11406404/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Gut microbiome (Cambridge, England)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1017/gmb.2023.11","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
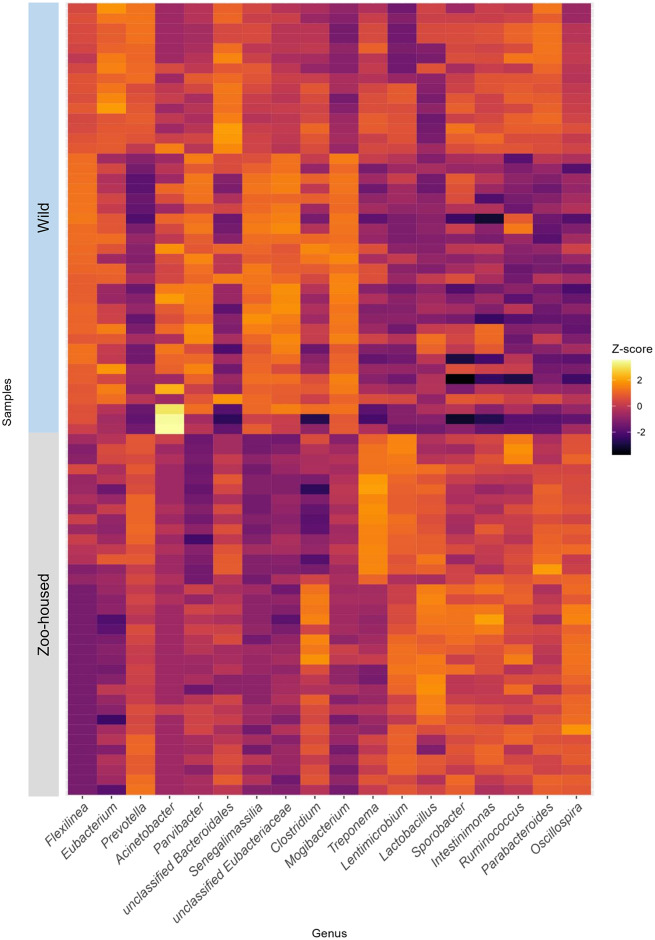
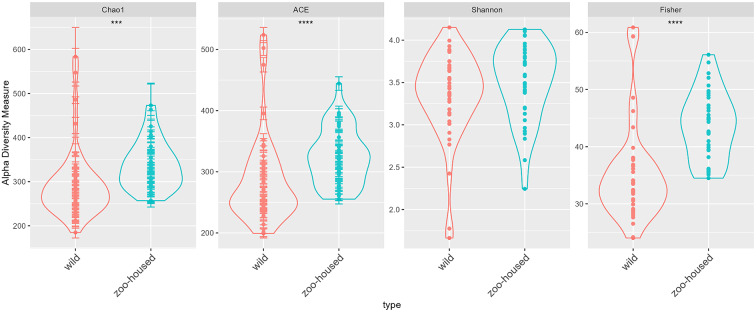
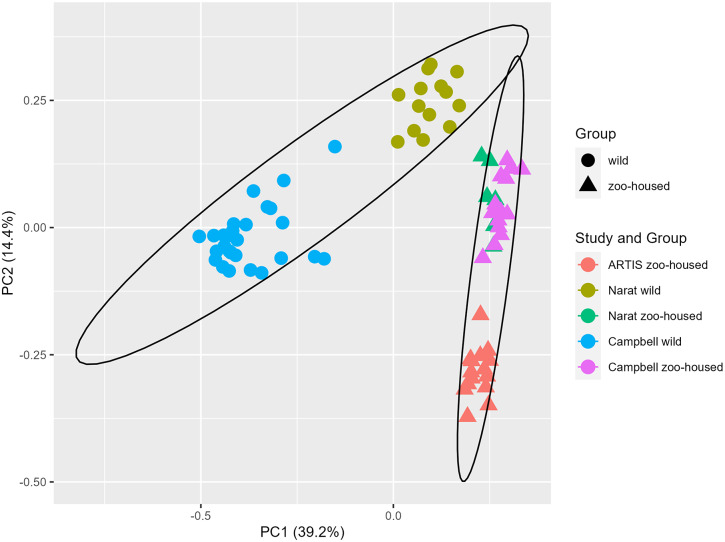
We carried out a comparative analysis between the bacterial microbiota composition of zoo-housed western lowland gorillas and their wild counterparts through 16S rRNA gene amplicon sequencing. In addition, we characterised the carbohydrate-active and methanogenic potential of the zoo-housed gorilla (ZHG) microbiome through shotgun metagenomics and RNA sequencing. The ZHG microbiota showed increased alpha diversity in terms of bacterial species richness and a distinct composition from that of the wild gorilla microbiota, including a loss of abundant fibre-degrading and hydrogenic Chloroflexi. Metagenomic analysis of the CAZyome indicated predominant oligosaccharide-degrading activity, while RNA sequencing revealed diverse cellulase and hemi-cellulase activities in the ZHG gut, contributing to a total of 268 identified carbohydrate-active enzymes. Metatranscriptome analysis revealed a substantial contribution of 38% of the transcripts from anaerobic fungi and archaea to the gorilla microbiome. This activity originates from cellulose-degrading and hydrogenic fungal species belonging to the class Neocallimastigomycetes, as well as from methylotrophic and hydrogenotrophic methanogenic archaea belonging to the classes Thermoplasmata and Methanobacteria, respectively. Our study shows the added value of RNA sequencing in a multiomics approach and highlights the contribution of eukaryotic and archaeal activities to the gut microbiome of gorillas.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


